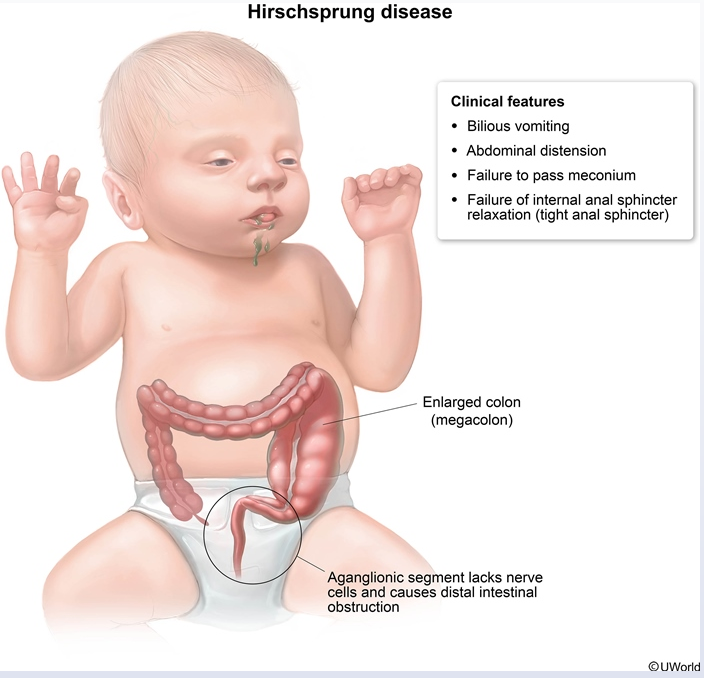
Hirschsprung
This child has signs of intestinal obstruction, including bilious vomiting, abdominal distension, hypoactive bowel sounds, and failure to pass meconium for the first 48 hours of life. These features, plus the explosive expulsion of gas and stool during rectal examination, are a classic presentation of Hirschsprung disease (HD), or congenital aganglionic megacolon. HD, which is usually diagnosed in the neonatal period, is due to failure of neural crest cells to migrate to the distal portion of the colon during development, causing functional obstruction.
However, milder cases may present later in life as chronic constipation, failure to thrive (decreased growth [<5th percentile]), distended abdomen, tight anal sphincter, and absence of stool in the rectal vault (as in this patient).
The gold standard for the diagnosis of HD is rectal suction biopsy. Rectal suction biopsy can be performed at the bedside without the need for general anesthesia and is very sensitive and specific in children age <3. Biopsy demonstrates the absence of ganglion cells and nerve fiber hypertrophy. Anorectal manometry, which measures pressure in the rectum and anal sphincter, is often used as to screen for HD; however, it is less accurate in infants age <1 month and lacks the sensitivity of suction biopsy (Choice B).
Treatment of HD is surgical resection of the aganglionic bowel segment and anastomosis of the normal bowel with the anus. Long-term improvements in quality of life are achieved in most patients, although some have lifelong continence problems. This is especially true in patients with underlying conditions such as trisomy 21.