cryoptogenic organizing pneumonia
- related: Pulmonology
- tags: #pulmonology
Organizing pneumonia is defined by histopathologic findings of patchy proliferation of granulation tissue that affects the terminal bronchiole, and alveolar ducts and spaces, and is associated with surrounding inflammation. This pattern often follows or is associated with various types of injury to the lung, including acute infection, radiation exposure, drug-induced pneumonitis, and autoimmune diseases. In patients in whom no cause is identified, the diagnosis is termed cryptogenic organizing pneumonia (COP).
Patients with COP typically present with cough, fever, and malaise for 6 to 8 weeks. Initial chest radiographs will demonstrate patchy opacities that mimic pneumonia and, as a result, patients are often initially misdiagnosed with community-acquired pneumonia and treated with standard antibiotics (Figure 7). However, nonresolving symptoms and failure to respond to antibiotics should raise suspicion for organizing pneumonia or COP. HRCT imaging will demonstrate ground-glass opacities or areas of alveolar consolidation resembling an infectious pneumonia, but findings can include peripheral nodules and nodules along the bronchovascular bundle. The diagnosis may not require lung biopsy if the clinical presentation and HRCT findings are consistent with COP. For cases with atypical presentation, lung biopsy may be necessary to make the diagnosis.
 Chest radiograph showing cryptogenic organizing pneumonia, demonstrating multiple patchy bilateral alveolar opacities that are nonspecific and may be difficult to distinguish from more typical infectious pneumonia. Infiltrates may be migratory, with resolution of established opacities as new areas appear on serial imaging. Imaging may also be nonspecific, showing interstitial infiltrates and alveolar opacification or one or more rounded nodules that may be interpreted as malignancy.
Chest radiograph showing cryptogenic organizing pneumonia, demonstrating multiple patchy bilateral alveolar opacities that are nonspecific and may be difficult to distinguish from more typical infectious pneumonia. Infiltrates may be migratory, with resolution of established opacities as new areas appear on serial imaging. Imaging may also be nonspecific, showing interstitial infiltrates and alveolar opacification or one or more rounded nodules that may be interpreted as malignancy.
Patients with COP typically respond to glucocorticoid therapy. In organizing pneumonia associated with an autoimmune disorder, treatment should focus on the autoimmune condition. Relapses of COP with tapering of glucocorticoids are common, and therefore a long taper of glucocorticoids or transition to alternate immunosuppressive therapy should be considered.
INTRODUCTION
Cryptogenic organizing pneumonia (COP), the idiopathic form of organizing pneumonia (formerly called bronchiolitis obliterans organizing pneumonia or BOOP), is a type of diffuse interstitial lung disease that affects the distal bronchioles, respiratory bronchioles, alveolar ducts, and alveolar walls. The primary area of injury is within the alveolar wall.
In addition to the cryptogenic form, secondary organizing pneumonia can be seen in association with connective tissue diseases, a variety of drugs, malignancy, and other interstitial pneumonias.
COP will be reviewed here. The other idiopathic interstitial pneumonias and an approach to the evaluation and diagnosis of interstitial lung disease in adults are discussed separately. (See "Idiopathic interstitial pneumonias: Classification and pathology" and "Approach to the adult with interstitial lung disease: Clinical evaluation" and "Approach to the adult with interstitial lung disease: Diagnostic testing".)
The American Thoracic Society (ATS) and European Respiratory Society (ERS) statement on the classification of idiopathic interstitial pneumonias, as well as other ATS guidelines, can be accessed through the ATS website at www.thoracic.org/statements.
EPIDEMIOLOGY
The exact incidence and prevalence of cryptogenic organizing pneumonia (COP) are unknown. A cumulative incidence of six to seven cases per 100,000 hospital admissions was found at a major teaching hospital in Canada, while in a 20 year review of national statistics for Iceland, the mean annual incidence was 1.1 per 100,000. Interstitial lung disease registry studies have reported a prevalence of COP of 5 percent in Greece and 10 percent in Spain.
PATHOGENESIS
The exact pathogenesis of cryptogenic organizing pneumonia (COP) remains unknown. It is thought that organizing pneumonia is a consequence of alveolar epithelial injury. The initial injury is followed by leakage of plasma proteins, recruitment of fibroblasts, and fibrin formation within the alveolar lumen.
Alveolar organization characterized by recruitment and proliferation of fibroblasts and myofibroblasts within the alveolar lumen form the fibroinflammatory buds (Masson’s polyps) characteristic of OP (picture 1). Abnormal regulation of vascular endothelial growth factor and matrix metalloproteinase has been reported in association with COP. However, dysregulation of these proteins has been observed in a large number of other pulmonary diseases, and their precise role in the pathogenesis of COP remains speculative. Regulation of angiogenesis and apoptosis may influence the reversibility of the fibrotic lesions in COP, compared to the irreversible lesions in usual interstitial pneumonia.
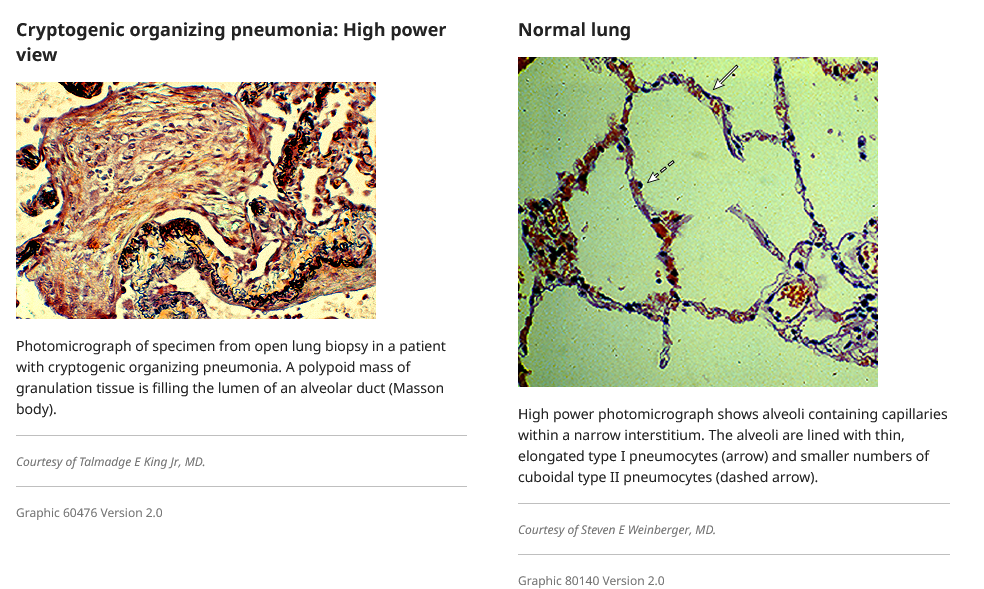
The inflammatory cells and fibrin deposits disappear and the intra-alveolar buds are remodeled into the interstitium leading to the restoration of the integrity and function of the alveolar unit.
Uncommonly, the lung parenchymal remodeling is characterized by residual interstitial inflammation and fibrosis throughout the alveolar wall in a pattern similar to nonspecific interstitial pneumonia (NSIP). A morphologic variant, cicatricial organizing pneumonia, with organizing airspace and airway granulation tissue leading to a dense mature eosinophilic scar tissue within the lumens of airways and airspaces, has been reported (picture 2).
A potential link between organizing pneumonia and microaspiration of gastric secretions in patients with gastroesophageal reflux disease (GERD) has been suggested. A link to environmental factors has also been suggested.
CLINICAL FEATURES
The disease onset is typically in the fifth or sixth decades of life, with males and females affected equally. Cryptogenic organizing pneumonia (COP) is only rarely reported in children. The reported frequency of current or past cigarette smoking ranges from 25 to 50 percent, making smoking unlikely to be a precipitating factor.
History — COP typically presents with cough, dyspnea, fever, and malaise that are of relatively short duration (eg, weeks to months). In almost three-fourths of the patients, symptoms are present for less than two months (figure 1). In one-half, the onset is heralded by the acute onset of a flu-like illness with fever, malaise, fatigue, and cough, but in other patients the onset of symptoms is less acute. In some patients, a lack of response to empiric antibiotics for community acquired pneumonia may be the initial clue to the presence of a noninfectious, inflammatory pneumonia.
The most common features of the history at presentation are:
- Persistent nonproductive cough (71 percent)
- Dyspnea (62 percent)
- Fever (44 percent)
- Malaise (48 percent)
- Weight loss of greater than 10 pounds (57 percent)
Hemoptysis is rarely reported as a presenting manifestation of COP.
As with all patients who present with interstitial lung disease, the history should include questions about any symptoms or history suggestive of connective tissue disease (eg, arthralgias, dry eyes, dry mouth, muscle weakness, numbness, tingling), current and recent medications, and history of exposure to therapeutic irradiation, fumes, or dusts. A list of diseases and medications that have been associated with secondary organizing pneumonia is provided in the table (table 1). A more complete list of drugs associated with secondary organizing pneumonia is available at www.pneumotox.com. (See "Approach to the adult with interstitial lung disease: Clinical evaluation", section on 'History'.)
Physical examination — In patients with COP, the physical examination often reveals inspiratory crackles (60 percent). Wheezing is rare and, when present, is heard in combination with crackles; clubbing is seen in less than 5 percent of cases. A normal pulmonary examination is found in less than 5 percent of patients at diagnosis.
Given the association of organizing pneumonia with many connective tissue diseases (table 1), the patient should be examined for extrapulmonary manifestations of these diseases such as alopecia, Gottron sign, heliotrope rash, muscle weakness or tenderness, sclerodactyly, and abnormal nailfold capillaroscopy. Identification of these features is described separately.
- (See "Approach to the adult with interstitial lung disease: Clinical evaluation", section on 'Physical examination'.)
- (See "Clinical manifestations of dermatomyositis and polymyositis in adults", section on 'Skin findings in dermatomyositis'.)
- (See "Clinical manifestations of rheumatoid arthritis", section on 'Symptoms and physical findings'.)
- (See "Clinical manifestations of dermatomyositis and polymyositis in adults", section on 'Muscle weakness'.)
- (See "Clinical manifestations and diagnosis of systemic sclerosis (scleroderma) in adults", section on 'Physical examination'.)
EVALUATION
The evaluation of patients with suspected cryptogenic organizing pneumonia (COP) usually begins with a chest radiograph obtained because of symptoms of dyspnea and/or cough. The chest radiographic appearance or a lack of clinical response to antibiotic therapy generally leads to additional studies to exclude atypical infections, malignancy, and systemic illnesses that might be associated with diffuse parenchymal lung disease. These studies typically include laboratory testing, high resolution computed tomography, pulmonary function testing, bronchoalveolar lavage, and transbronchoscopic or surgical lung biopsy. The evaluation of interstitial lung disease is discussed separately. (See "Approach to the adult with interstitial lung disease: Clinical evaluation" and "Approach to the adult with interstitial lung disease: Diagnostic testing".)
Laboratory tests — No laboratory studies are specific for COP. However, in the evaluation of patients with dyspnea, cough, fever, and pulmonary radiographic opacities, the usual laboratory tests include a complete blood count and differential, blood urea nitrogen, creatinine, hepatic function tests, and urinalysis. (See "Approach to the adult with interstitial lung disease: Diagnostic testing", section on 'Laboratory tests'.)
Leukocytosis is present in about 50 percent of patients with COP. An elevated initial erythrocyte sedimentation rate (ESR) (frequently reaching or exceeding 100 mm) and a positive C-reactive protein are each observed in 70 to 80 percent, although we do not routinely obtain these tests.
For hospitalized patients whose clinical presentation suggests community acquired pneumonia, additional tests are usually obtained, such as blood cultures, sputum gram stain, sputum enzyme immunoassay (EIA), immunofluorescence, or polymerase chain reaction (PCR) for respiratory viruses, urinary studies for pneumococcal and Legionella antigens, and HIV testing.
As organizing pneumonia may also occur in association with various connective tissue diseases, testing for these diseases (eg, antinuclear antibody, rheumatoid factor, creatine kinase, anti-topoisomerase [anti-Scl70], anti-centromere antibody, anti-double-stranded DNA, anti-JO1) is appropriate when a connective tissue disease is suspected on the basis of symptoms or signs (table 1). In patients with COP (ie, without concomitant connective tissue disease), autoantibodies are usually negative or present in very low titer. Laboratory testing to identify underlying connective tissues disease in the setting of interstitial lung disease is discussed separately (algorithm 1). (See "Approach to the adult with interstitial lung disease: Diagnostic testing", section on 'Laboratory tests'.)
Chest imaging studies
Chest radiograph — A chest radiograph is obtained in virtually all adults with persistent symptoms of dyspnea and cough, particularly in the presence of fever. The chest radiograph manifestations of COP are typically quite distinctive with bilateral, patchy or diffuse, consolidative or ground glass opacities in the presence of normal lung volumes (image 1 and image 2). Other findings may include:
- A peripheral distribution of the opacities, similar to that seen in chronic eosinophilic pneumonia (image 1). (See "Overview of pulmonary eosinophilia", section on 'Chronic eosinophilic pneumonia'.)
- Recurrent or migratory pulmonary opacities are common (up to 50 percent of subjects in some series).
- Rarely, unilateral consolidative and ground-glass opacities.
- Rarely, irregular linear or nodular opacities as the only radiographic manifestation (image 3).
- Other rare radiographic abnormalities include pleural effusion, pleural thickening, hyperinflation, and cavities.
Computed tomographic scanning — High resolution computed tomography (HRCT) lung scans are typically obtained to evaluate abnormalities seen on conventional chest radiographs. Rarely, a patient may have dyspnea and hypoxemia or a reduced diffusing capacity (DLCO) without a definite abnormality on chest radiograph, and an HRCT is obtained to evaluate the gas transfer abnormality. HRCT results are also used to guide site selection for patients undergoing bronchoalveolar lavage or lung biopsy.
HRCTs from patients with COP often reveal more extensive disease than expected from review of the plain chest radiograph. Radiographic patterns include patchy air-space consolidation, ground-glass opacities, small nodular opacities, and bronchial wall thickening with dilation (image 4). Patchy opacities occur more frequently in the periphery of the lung and are often in the lower lung zone.
Less common radiographic appearances include multiple nodules or masses that may cavitate, micronodules, irregular reticular opacities in a subpleural location, and crescentic or ring-shaped opacities. Rarely, a single nodule is the only manifestation of organizing pneumonia; this is termed focal organizing pneumonia (image 5). A retrospective study of 12 patients with biopsy-proven COP with a solitary lung mass showed that all lesions were subpleural, and in six the masses had a heterogeneous density; among these, four masses were cavitary. Lymphadenopathy was seen in seven patients. In a case series, mediastinal lymphadenopathy (diameter ≥10 mm in short axis) was reported in 6 of 16 patients with COP. Pleural effusion is uncommon.
Confident radiographic differentiation of COP from chronic eosinophilic pneumonia is frequently NOT possible, as both diseases have a predilection for the peripheral lung fields (image 6 and image 7). (See 'Differential diagnosis' below and "Overview of pulmonary eosinophilia", section on 'Chronic eosinophilic pneumonia'.)
Pulmonary function tests — Pulmonary function tests are obtained in most ambulatory adults with dyspnea and cough to determine whether the symptoms are associated with an obstructive or restrictive impairment and whether a gas transfer defect is present. The severity of the impairment is one of the factors used to determine the need for invasive diagnostic testing. (See "Approach to the adult with interstitial lung disease: Diagnostic testing", section on 'Pulmonary function testing' and "Overview of pulmonary function testing in adults".)
A mild to moderate restrictive ventilatory defect is the most common finding in COP (figure 2). However, an obstructive defect (forced expiratory volume in one second/forced vital capacity [FEV1/FVC] ratio less than 70 percent) was found in 20 percent of patients in one report, all of whom were current or former smokers. Lung volumes and flow measurements are occasionally normal.
Gas exchange abnormalities are extremely common. The DLCO is reduced in the majority of patients. Resting and/or exercise arterial hypoxemia (each defined by an alveolar-arterial oxygen gradient greater than 20 mmHg) are present in more than 80 percent of subjects. Pulse oxygen saturation (SpO2) may be normal or reduced at rest, but commonly is decreased with exertion.
Flexible bronchoscopy — Flexible bronchoscopy is usually performed to obtain bronchoalveolar lavage (BAL) samples to evaluate for infection, hemorrhage, and malignancy. A transbronchial biopsy is sometimes performed during the procedure to obtain tissue for histopathologic and microbiologic studies, although the size of the samples is generally not large enough to make a definitive diagnosis of COP. Alternatively, the evaluation may proceed directly to surgical lung biopsy when infection seems unlikely or the diagnostic process needs to be expedited.
Bronchoalveolar lavage — In patients with COP, bronchoalveolar lavage (BAL) findings are nonspecific, so the main role for BAL is to identify processes that are in the differential diagnosis of subacute onset of cough, dyspnea, and radiographic opacities, such as alveolar hemorrhage, malignancy, or opportunistic infection. The technique of BAL and the preparation of lavage samples are discussed separately. (See "Role of bronchoalveolar lavage in diagnosis of interstitial lung disease" and "Basic principles and technique of bronchoalveolar lavage".)
The location for BAL may be chosen based on examination of the chest HRCT to select an area with radiographic evidence of involvement. Alternatively, in diffuse disease, the right middle lobe or lingula is lavaged most commonly to optimize fluid recovery. (See "Basic principles and technique of bronchoalveolar lavage", section on 'Optimal site'.)
BAL findings have been reported in a limited number of patients with COP. The percentage of the volume of BAL fluid recovered from patients with COP is less than that recovered from healthy volunteers, but the total number of cells recovered is greater. Typical BAL findings include increases in lymphocytes (20 to 40 percent), neutrophils (5 to 10 percent), and eosinophils (5 to 25 percent) with the level of lymphocytes being higher than that of eosinophils. This "mixed pattern" of increased cellularity is thought to be characteristic, although not diagnostic of COP. Patients with COP tend to have higher lymphocyte counts than do those with idiopathic pulmonary fibrosis, but similar in proportion to hypersensitivity pneumonitis. BAL neutrophilia (defined as the presence of 10 percent or more neutrophils) tends to correlate with relapse of OP. (See "Role of bronchoalveolar lavage in diagnosis of interstitial lung disease", section on 'Mixed cellularity BAL'.)
Other (nondiagnostic) BAL abnormalities that may be found in COP patients include foamy macrophages, mast cells, plasma cells, a decreased CD4/CD8 T cell ratio, an increase in activated T lymphocytes based on HLA-DR or interleukin-2 receptor expression. In research studies, increased levels of Th1 related cytokines, including interferon (IFN)-y, interleukin (IL)-12 and IL-18, have been reported, although these tests are not used clinically.
Transbronchial lung biopsy — The main role for transbronchial biopsy in patients with suspected COP is to identify other disease processes in the differential diagnosis, because the small size of transbronchial lung biopsies is often inadequate for definitive confirmation of COP and exclusion of other concomitant processes. The histologic features of organizing pneumonia can be seen as a minor finding in other interstitial lung diseases, so reliance on small transbronchial biopsies increases the likelihood of missing the central diagnosis. On the other hand, some guidelines suggest that a diagnosis can be made on the basis of a transbronchial biopsy in a patient with typical clinical and radiologic features. (See 'Histopathology' below and 'Differential diagnosis' below.)
The location for performing transbronchial biopsies is chosen based on examination of the chest HRCT, which allows selection of an area with radiographic evidence of involvement. Transbronchial biopsy samples are sent for histopathologic analysis and microbiologic stains and cultures. (See "Role of lung biopsy in the diagnosis of interstitial lung disease", section on 'Transbronchial lung biopsy' and "Flexible bronchoscopy in adults: Overview" and "Flexible bronchoscopy in adults: Associated diagnostic and therapeutic procedures", section on 'Transbronchial biopsy'.)
In the appropriate clinical setting, CT-guided lung biopsy may yield histologic features of organizing pneumonia reducing the need for surgical lung biopsy.
Surgical lung biopsy — An open or thoracoscopic lung biopsy is generally needed to obtain an adequate sample of lung tissue (eg, >4 cm diameter in the greatest dimension when inflated) for definitive diagnosis. The location of the lung biopsy is typically chosen based on areas of abnormality identified on the HRCT scan and on the accessibility of these areas to the surgical approach. Samples are sent for histopathologic and microbiologic analysis. (See "Role of lung biopsy in the diagnosis of interstitial lung disease", section on 'Surgical lung biopsy'.)
In addition to an adequate sample of tissue, it is important that the pathologist be given adequate clinical information to guide the search for the specific lesions and patterns that support the diagnosis. (See 'Histopathology' below and "Interpretation of lung biopsy results in interstitial lung disease", section on 'Multidisciplinary discussion'.)
HISTOPATHOLOGY
The histopathologic lesions characteristic of cryptogenic organizing pneumonia (COP) include excessive proliferation of granulation tissue, which consists of loose collagen-embedded fibroblasts and myofibroblasts, involving alveolar ducts and alveoli, with or without bronchiolar intraluminal polyps. Intraluminal plugs of granulation tissue may extend from one alveolus to the adjacent one through the pores of Kohn, giving rise to the characteristic "butterfly" pattern (picture 1 and picture 3). It is thought that bronchiolar lesions reflect extension of intraluminal plugs of granulation tissue from alveolar sacs and ducts to the bronchioles. Other features include a uniform appearance within involved areas and a patchy distribution without severe disruption of the lung architecture. While the pathologic lesion is predominantly within the airspace, mild chronic inflammation (eg, lymphocytes and plasma cells) is present in the alveolar walls (picture 4).
There are, in addition, other key histopathologic features that are listed in the table (table 2).
Histologic features of organizing pneumonia (OP) can also be seen in combination with other idiopathic interstitial pneumonias (table 3). However, the portion of changes suggestive of OP is small (<10 percent) relative to the characteristic changes of these other interstitial pneumonias. (See "Interpretation of lung biopsy results in interstitial lung disease", section on 'Interstitial pneumonias'.)
DIAGNOSIS
Cryptogenic organizing pneumonia — The diagnosis of cryptogenic organizing pneumonia (COP) depends upon demonstration of the typical histopathologic features in a patient with a compatible clinical and radiographic pattern in the absence of a contributing factor or disease process (table 1). When making a histopathologic diagnosis of COP, it is necessary to satisfy two key criteria:
- The presence of the characteristic histopathologic features of organizing pneumonia (OP), including intraluminal plugs of inflammatory debris predominantly within small airways, alveolar ducts, and adjacent alveoli and also mild interstitial inflammation in the surrounding lung (table 2). The "plugs" of inflammatory debris consist of granulation tissue with fibroblasts and myofibroblasts embedded in a connective matrix. (See 'Histopathology' above.)
- The absence of histopathologic features suggestive of another process (eg, poorly formed granulomas suggestive of hypersensitivity pneumonitis, prominent eosinophilia suggestive of chronic eosinophilic pneumonia, temporal heterogeneity of the lesions and fibroblast foci suggestive of usual interstitial pneumonitis) (table 1). (See 'Differential diagnosis' below.)
In order to have enough tissue for the pathologist to exclude other processes, such as nonspecific interstitial pneumonia or usual interstitial pneumonia, we prefer to obtain a lung biopsy via video-assisted thoracoscopic surgery (VATS) or open thoracotomy rather than transbronchial biopsy. (See 'Transbronchial lung biopsy' above and 'Surgical lung biopsy' above.)
Excluding other etiologies of organizing pneumonia — When the diagnosis of OP is made by lung biopsy, the next step is to determine whether the disease is cryptogenic or secondary to another process. It is impossible to differentiate COP from secondary OP based on radiologic or pathologic findings. Thus, a careful review of the patient’s history, physical examination, medication usage, potential exposures, and underlying diseases is needed to determine whether COP is indeed cryptogenic. Potential disease and drug associations with organizing pneumonia are listed in the table (table 1).
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of cryptogenic organizing pneumonia (COP) includes a broad spectrum of diseases that have similar clinical features, a similar radiographic appearance, similar histopathologic findings, or overlapping histopathologic features.
-
Community acquired pneumonia – The onset of symptoms and radiographic appearance of COP often mimic community acquired pneumonia, but the persistence of symptoms and lack of response to antibiotics suggests COP rather than bacterial or viral pneumonia. As organizing pneumonia can be a consequence of certain infections, positive culture results do not necessarily exclude the diagnosis (table 1). (See "Clinical evaluation and diagnostic testing for community-acquired pneumonia in adults".)
-
Idiopathic interstitial pneumonias – COP is one type of idiopathic interstitial pneumonia (IIP), and the other types of IIP are in the differential of COP (table 3). Sometimes small areas (<10 percent) of organizing pneumonia are seen within a broader appearance of another IIP, such as nonspecific interstitial pneumonia or idiopathic pulmonary fibrosis (table 1). Clinicopathologic collaboration is needed to determine the correct diagnosis. (See 'Histopathology' above and "Interpretation of lung biopsy results in interstitial lung disease", section on 'Multidisciplinary discussion' and "Interpretation of lung biopsy results in interstitial lung disease", section on 'Interstitial pneumonias' and "Idiopathic interstitial pneumonias: Classification and pathology".)
-
Hypersensitivity pneumonitis – The clinical presentation of COP is similar to that of subacute hypersensitivity pneumonitis (cough, dyspnea, fatigue, anorexia, and weight loss). The radiographic findings typical of subacute hypersensitivity pneumonitis (eg, diffuse micronodules, ground glass attenuation) are also seen in some patients with COP. A known exposure to an etiologic agent, a higher percentage of lymphocytes on bronchoalveolar lavage, and the presence of poorly formed granulomas on lung biopsy all favor hypersensitivity pneumonitis. However, small areas of organizing pneumonia can be seen in hypersensitivity pneumonitis, so differentiation of these diseases requires clinicopathologic collaboration (table 1). (See "Hypersensitivity pneumonitis (extrinsic allergic alveolitis): Clinical manifestations and diagnosis", section on 'Histopathology' and "Interpretation of lung biopsy results in interstitial lung disease", section on 'Multidisciplinary discussion'.)
-
Granulomatous organizing pneumonia – Granulomatous organizing pneumonia (GOP) is a newly described variant of COP, characterized by a histopathologic pattern of organizing pneumonia in close association with small poorly formed non-necrotizing granulomas. These non-necrotizing granulomas must be confined to the same peribronchiolar location and within the organizing pneumonia pattern. The cause of this pattern is not known, but it is thought to be an allergic or immunologic response.
-
Chronic eosinophilic pneumonia – Chronic eosinophilic pneumonia (CEP) can have a similar clinical presentation to COP, and the radiographic pattern of subpleural patchy areas of consolidation can be seen in both of these diseases. Furthermore, a mixed picture that includes features of organizing pneumonia and eosinophilic pneumonia may be seen, suggesting that CEP can transition to COP in some patients. The differentiation of these processes usually depends on the relative proportions of the histopathologic patterns and the response to systemic glucocorticoids. COP generally has a slower response over several days to a week, while chronic eosinophilic pneumonia typically has a more rapid response (hours to a couple of days). (See "Overview of pulmonary eosinophilia", section on 'Chronic eosinophilic pneumonia' and "Interpretation of lung biopsy results in interstitial lung disease", section on 'Eosinophilic pneumonia' and "Chronic eosinophilic pneumonia", section on 'Assessing the response to therapy'.)
-
Pulmonary lymphoma – The radiographic appearance of pulmonary lymphoma can be similar to COP with patchy, nodular areas of consolidation and an air bronchogram. A single area of consolidation is more common in lymphoma than in COP, although focal COP can be seen. The diseases are differentiated by the histopathologic and immunohistochemical appearance. (See "Clinical manifestations, pathologic features, and diagnosis of extranodal marginal zone lymphoma of mucosa associated lymphoid tissue (MALT)".)
Pulmonary lymphomatoid granulomatosis (PLG) is a form of lymphoproliferative disease that is in the differential of COP and pulmonary lymphoma. In PLG, multiple poorly defined nodules are typically present on chest computed tomography and lung biopsy shows the triad of polymorphic lymphoid infiltrates, transmural infiltration of arteries and veins by lymphoid cells ("angiitis"), and focal areas of necrosis within the lymphoid infiltrates. (See "Pulmonary lymphomatoid granulomatosis", section on 'Pathology'.)
-
Diffuse alveolar damage – Diffuse alveolar damage (DAD) is a nonspecific histopathologic reaction to lung injury, most commonly seen in patients with the acute respiratory distress syndrome (ARDS). Occasional cases of COP have a fulminant onset and progress rapidly to respiratory failure. In these patients, COP is the dominant pattern with areas of DAD. Conversely, a component of OP may be seen on lung biopsy specimens obtained during the proliferative phase of ARDS due to other causes (eg, acute interstitial pneumonia). Careful review of the histopathologic findings is needed to differentiate these processes. (See "Acute interstitial pneumonia (Hamman-Rich syndrome)", section on 'Pathology' and "Acute respiratory distress syndrome: Epidemiology, pathophysiology, pathology, and etiology in adults", section on 'Pathologic stages'.)
-
Acute fibrinous and organizing pneumonia – Acute fibrinous and organizing pneumonia (AFOP) is associated with a clinical picture of acute lung injury and can be idiopathic or associated with other processes such as hypersensitivity pneumonitis, infection, drug toxicity, eosinophilic pneumonia, and rheumatic disease. The histopathology is characterized by intra-alveolar fibrin deposition (fibrin "balls") and associated organizing pneumonia, but without hyaline membranes (picture 5). Patients with multifocal fibrin deposits or acute fibrinous and organizing pneumonia on lung biopsies experience a higher rate of OP relapse compared to those with no or focal fibrin (60 versus 6 percent, p<0.05). (See "Interpretation of lung biopsy results in interstitial lung disease", section on 'Rare histopathologic interstitial pneumonia patterns'.)
TREATMENT
The treatment of cryptogenic organizing pneumonia (COP) has not been studied in randomized trials, so treatment decisions are based on practice guidelines, observations from case series, and our clinical experience. The decision to initiate therapy and the choice of initial therapy depend on the severity of symptoms and pulmonary function impairment at presentation, the radiographic extent of disease, and the rapidity of progression..
Mild stable disease — For patients with minimal symptoms, near normal or normal pulmonary function tests, and mild radiographic involvement, it is reasonable to monitor without therapy pending any worsening of symptoms or pulmonary function. Among such patients with mild disease, spontaneous remission may occasionally occur. Patients are reassessed at 8 to 12 week intervals regarding any increase in symptoms or worsening of pulmonary function.
For patients with mild to moderate disease, macrolides may be an option, particularly for patients with normal lung function who prefer to avoid glucocorticoid therapy. A few case reports have described responses to a macrolide antibiotic (eg, clarithromycin 250 to 500 mg twice a day) in patients with mild symptoms. In these cases, a prolonged course of three to six months was needed; tapering of the macrolide to a once daily dose was successful in some patients, but early discontinuation often led to recurrent disease. However, macrolide therapy appears less effective compared with glucocorticoids. In a long-term retrospective study of 87 patients with biopsy-proven COP, 11 patients received macrolides (average duration 4.2 months, range 3 to 12 months); 7 of them achieved remission without relapse, while 4 did not achieve remission.
It is thought that the benefit of the macrolide is related to anti-inflammatory rather than antimicrobial effects. However, the diagnosis of COP is often made after a patient has failed one or more courses of a macrolide antibiotic for a presumed community acquired pneumonia, in which case it is unlikely that macrolide therapy will be sufficient.
Persistent or gradually worsening disease — The majority of patients with COP have persistent, bothersome, and progressive symptoms associated with moderate pulmonary function test impairment and diffuse radiographic changes. For these patients, we suggest initial therapy with oral glucocorticoids, which are usually associated with rapid improvement. In 12 studies with a total of 160 patients, a complete response to prednisone was seen in 60 percent, partial response in 27 percent, nonresponse in 14 percent, and a fatal outcome in 6 percent. The optimal initial dose of systemic glucocorticoid therapy is not known. We typically use an initial dose of prednisone of 0.75 to 1 mg/kg per day, using ideal body weight (calculator 1), to a maximum of 100 mg/day given as a single oral dose in the morning, as suggested by the British Thoracic Society guidelines. In our practice, the initial dose for most patients is 60 mg per day.
We suggest maintaining the initial oral dose for four to eight weeks. If the patient is stable or improved, the prednisone dose is gradually tapered to 0.5 to 0.75 mg/kg per day (using ideal body weight) for the ensuing four to six weeks. After three to six months of oral prednisone, the dose is gradually tapered to zero if the patient remains stable or improved.
In addition to clinical assessment, the patient should be routinely followed with a conventional chest radiograph and pulmonary function testing every two to three months as long as systemic glucocorticoid therapy is required. At the first sign of worsening or recurrent disease, the prednisone dose should be increased to the prior dose or reinstituted promptly. Importantly, the chest radiograph may change before the patient develops significant symptoms. After cessation of glucocorticoids, we follow the patient clinically for the next year and repeat the chest radiograph approximately every three months.
Glucocorticoid treatment is associated with a variety of side effects. Thus, there needs to be a careful and ongoing assessment of risks and benefits. Steps to minimize the adverse effects (eg, infection, osteoporosis, myopathy, adrenal suppression) of systemic glucocorticoids are discussed separately. (See "Major side effects of systemic glucocorticoids" and "Prevention and treatment of glucocorticoid-induced osteoporosis" and "Pharmacologic use of glucocorticoids", section on 'Minimizing glucocorticoid side effects' and "Glucocorticoid withdrawal", section on 'Recommended tapering regimen'.)
Failure to respond to systemic glucocorticoids — For patients who have stable disease but fail to improve with systemic glucocorticoids, we review the initial diagnostic testing results to be sure that the patient does not have an alternate diagnosis. (See 'Differential diagnosis' above.)
Superimposed infection is also excluded, particularly if deterioration has followed an initial improvement. In this setting, a repeat bronchoscopy with bronchoalveolar lavage is often indicated.
-
Cytotoxic therapy − Except in very rare cases where other approaches have failed, we do not recommend the use of cytotoxic agents (eg, azathioprine or cyclophosphamide). After reconfirming the diagnosis of COP, a cytotoxic agent is usually started while maintaining oral prednisone. We typically use cyclophosphamide, although data in support of this choice are limited to case reports. Some experts prefer azathioprine because of its lower side effect profile. For patients with normal renal function, the initial dose is 1 to 2 mg/kg per day (given as a single daily dose) up to a maximum of 150 mg/day, although the optimal dose in COP is unknown. We usually start at 50 mg daily and slowly increase the dose over two to four weeks. For patients with abnormal renal function, the dose of azathioprine or cyclophosphamide is modified (table 4), or an alternate agent is used. A trial of at least three months is needed to ensure an adequate opportunity for clinical response. Due to the toxicity of cyclophosphamide, we discontinue this agent after six months. Potential adverse effects of cyclophosphamide and guidance for its safe administration are discussed separately. (See "General toxicity of cyclophosphamide in rheumatic diseases" and "General principles of the use of cyclophosphamide in rheumatic diseases".)
-
Other therapies − The addition of a macrolide antibiotic to systemic glucocorticoid therapy has been beneficial in case reports. In a case series, four patients who experienced relapse of their disease following discontinuation of glucocorticoids were successfully treated with macrolides, with no apparent ill effects. For patients with recurrent disease, a literature review suggested that macrolides be used as adjunctive therapy with other treatments, such as a glucocorticoid.
Use of oral mycophenolate mofetil, an inhibitor of proliferating lymphocytes, has been recommended as a steroid-sparing agent in the treatment of COP.
In a few case reports, cyclosporine was used in combination with glucocorticoids to treat rapidly progressive disease. A case series described four patients who had failed to respond to glucocorticoids, but experienced at least some improvement with rituximab added to glucocorticoids; one patient had a complete response. Intravenous immunoglobulins have been used in steroid-resistant OP.
Inability to taper glucocorticoids or intolerance of adverse effects — Relapses are common and usually occur when the glucocorticoids are tapered after one to three months of treatment. In some series, over one-half of patients experience at least one clinical relapse during the course of their disease. Most of these patients will improve when retreated with glucocorticoids, and the occurrence of relapses generally does not appear to affect the overall long-term prognosis.
Patients with persistent or frequently recurrent (more than three) episodes may require long-term treatment with prednisone and a glucocorticoid-sparing agent. We typically use azathioprine as a glucocorticoid-sparing agent in patients who have responded to glucocorticoid therapy, but require a high dose to control their disease, and those who are intolerant of glucocorticoid adverse effects. However, data in support of glucocorticoid-sparing agents for COP are limited. A few case reports have described a favorable outcome with a combination of azathioprine and low dose prednisone.
Toxicity of azathioprine is partly related to deficiency in the enzyme thiopurine methyltransferase (TPMT), and analysis of the TPMT gene prior to the administration of azathioprine may help predict those individuals at risk for severe toxicity. The pharmacology and adverse effects of azathioprine are discussed separately. (See "Pharmacology and side effects of azathioprine when used in rheumatic diseases".)
Fulminant disease — For the minority of patients who present with rapidly progressive and extensive disease (eg, requiring high flow supplemental oxygen), after exclusion of infection, we suggest initial therapy with intravenous glucocorticoids (eg, methylprednisolone 125 to 250 mg every six hours or a pulse of 750 to 1000 mg given once daily for three to five days) based on clinical experience and case reports. Once the patient shows signs of improvement (usually within five days), glucocorticoid therapy is transitioned to oral prednisone at a dose of 0.75 to 1 mg/kg per day (using ideal body weight) to a maximum of 100 mg/day. Subsequent tapering and prevention of long-term adverse effects of glucocorticoid therapy are described above. (See 'Persistent or gradually worsening disease' above.)
It is unknown whether a second immunosuppressive agent should be added in patients who require mechanical ventilation for respiratory failure or do not respond rapidly to intravenous glucocorticoids, but we generally add a second agent. We typically choose cyclophosphamide, given the greater experience with it. Cyclophosphamide is discontinued after six months or sooner if the patient has adverse effects. (See 'Failure to respond to systemic glucocorticoids' above and "General principles of the use of cyclophosphamide in rheumatic diseases".)
Focal organizing pneumonia — Resection of a solitary lung nodule containing focal organizing pneumonia is adequate initial therapy for most patients. In two retrospective studies, focal organizing pneumonia was found in resected solitary pulmonary nodules in a combined total of 43 patients. No further therapy was given and there was no recurrence in 41 patients; the two patients with local recurrences were treated successfully with glucocorticoids. A subsequent report further endorses that surgical resection alone is sufficient for the management of focal organizing pneumonia.
PROGNOSIS
Recovery, usually with complete clinical and physiologic improvement and normalization of the chest film, occurs in two-thirds of patients treated with glucocorticoids (figure 3). Symptomatic improvement is occasionally quite dramatic, occurring in one to two weeks, although most patients improve more gradually over several weeks to a few months. Serial high resolution computed tomography (HRCT) scans may reveal persistent abnormalities despite clinical improvement. Patients with airspace opacities on chest radiograph have a much better outcome than those with reticular opacities. Approximately one-third of patients experience persistent symptoms, abnormalities on pulmonary function testing, and radiographic disease. In general, the severity of illness at presentation is related to potential for subsequent relapse, eg, severity of hypoxemia, extent of radiographic abnormalities, severity of the decline in lung function, presence of an underlying illness, or length of delay in beginning treatment.
The overall prognosis of COP is much better than that of other interstitial lung diseases, such as idiopathic pulmonary fibrosis, fibrosis nonspecific interstitial pneumonia, and acute interstitial pneumonia. Rapidly fatal COP is uncommon.