primary biliary cholangitis
- related: GI, PSC primary sclerosing cholangitis
- tags: #GI
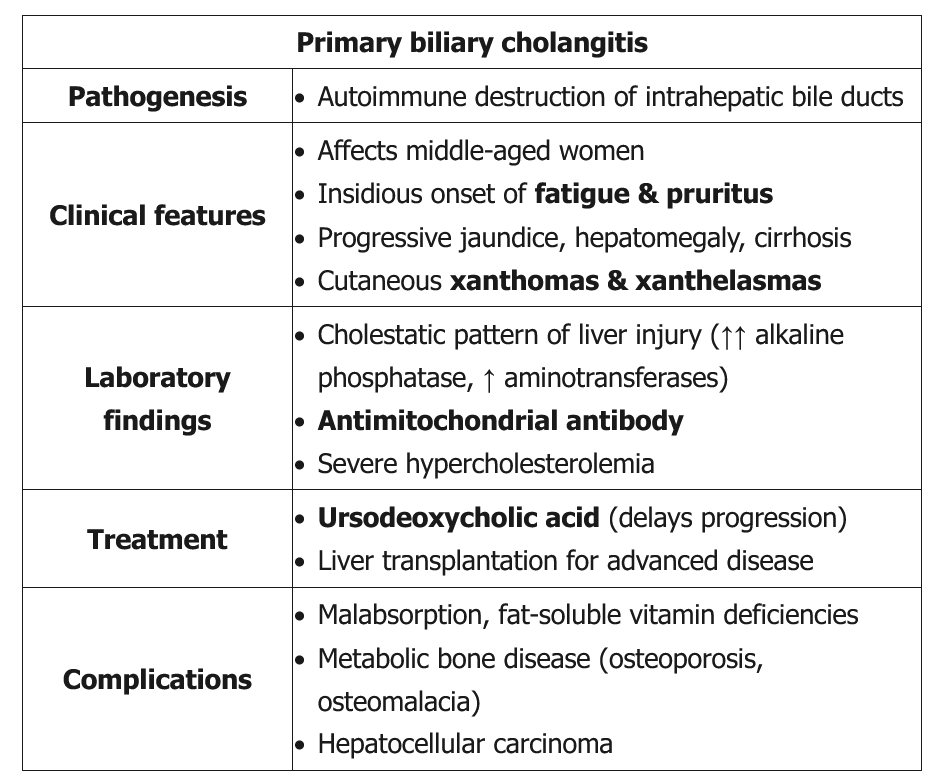
Primary biliary cholangitis (PBC), previously termed primary biliary cirrhosis, is an autoimmune disease affecting the small and medium bile ducts with a female to male predominance of 9 to 1. It is more common in individuals of European descent. PBC can present with fatigue and pruritus, but many patients are asymptomatic, with the diagnosis suggested by elevated ALP levels. Diagnosis does not require liver biopsy when the ALP is at least 1.5 times the upper limit of normal and antimitochondrial antibody (AMA) testing is positive, or when other PBC-specific autoantibodies, including sp100 or gp210, are present if antimitochondrial antibody testing is negative. In patients with negative antibody test results and strong suspicion for PBC, a liver biopsy is necessary. Transient or MRI elastography can be used for fibrosis staging.
The initial treatment is ursodeoxycholic acid. Response to treatment is defined by improvement of ALP level to less than 1.67 times the upper limit of normal. Patients whose disease does not respond to ursodeoxycholic acid should receive obeticholic acid. Dose reductions are required for obeticholic acid use in patients with decompensated cirrhosis to avoid worsening liver failure. Ursodeoxycholic acid treatment results in histologic improvement, better survival rates, and diminished need for liver transplantation. Patients who present with normal bilirubin and albumin levels and respond to treatment have a life expectancy similar to that of individuals without PBC.
PBC is associated with other autoimmune conditions, particularly autoimmune thyroid disease. Therefore, in patients with PBC, thyroid-stimulating hormone level should be checked on a yearly basis. In patients with a PBC score of 4.1 or greater, upper endoscopy is indicated to assess for esophageal varices. First-degree relatives of patients with PBC, especially women, should be screened by checking their ALP level periodically. Patients with advanced disease should be managed like other patients with cirrhosis and portal hypertension (see Complications of Advanced Liver Disease). In addition, the American Association for the Study of Liver Diseases recommends screening for hepatocellular carcinoma with ultrasonography at 6-month intervals for men and patients with cirrhosis. Patients should ensure dietary intake of 1000 to 1500 mg of calcium and 1000 IU of vitamin D daily, with supplements if needed. Patients are at increased risk for osteoporosis; appropriate screening and treatment with bisphosphonate therapy should be considered. Patients with elevated lipid levels may be at risk for cardiovascular disease and can be considered for lipid-lowering therapy. Fat-soluble vitamin deficiencies should be treated with parenteral or water-soluble supplements. Liver transplant outcomes for patients with PBC are excellent, with a 1-year survival rate greater than 90% and a recurrence rate of approximately 20% at 5 years after liver transplantation.
Up to 50% of patients develop metabolic bone disease, which may manifest as osteoporosis and/or osteomalacia. The cause of osteoporosis in PBC is unknown but is thought to be due to cholestasis leading to an accumulation of substances that inhibit bony turnover. Additionally, the disease predominately affects middle aged female patients who are already at risk for osteoporosis. Patients may have malabsorption of fat-soluble vitamins, but those with PBC-related osteoporosis usually have normal vitamin D levels. Nevertheless, patients with PBC should receive routine supplementation with calcium and vitamin D in addition to regular bone density screening.