Other Rheumatologic Diseases
- related: Rheumatology
- tags: #rheumatology
Behçet Syndrome
Behçet syndrome is a systemic disease associated with inflammatory infiltration of multiple organs, as well as small-vessel vasculitis and large-vessel vasculopathy. The characteristic clinical features are recurrent painful oral and genital mucosal ulcerations (Figure 29) and inflammatory eye disease (panuveitis, retinal vasculitis). A characteristic clinical finding is pathergy, an inflamed papular or pustular response to local skin injury that is clinically defined by similar lesions appearing 48 hours after skin prick with a sterile needle (Figure 30). Other clinical manifestations include venous thrombosis that may affect the large veins, including the vena cava and the dural venous sinuses. Central nervous system (CNS) manifestations include brainstem lesions and aseptic meningitis; the most common CNS symptoms are headache and diplopia. Inflammatory arthritis (usually in the knees), skin lesions, and gastrointestinal inflammation/ulceration indistinguishable from inflammatory bowel disease also occur. Diagnostic criteria are listed in Table 42.
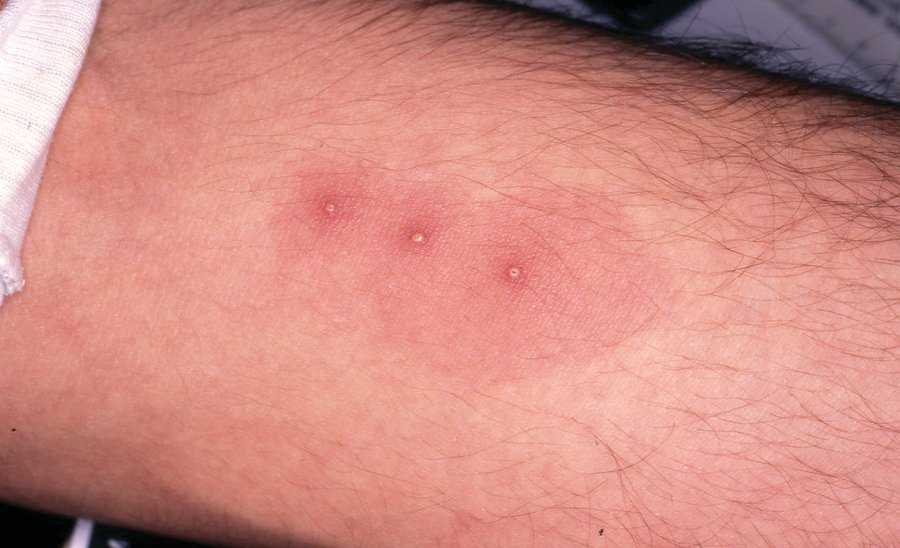
Behçet syndrome is most common in Asia, with the highest reported prevalence in Turkey; it is much less common in North America. It is slightly more prevalent in men than women, and men usually have more severe disease. HLA-B51 is strongly associated with the disease and is seen in patients with varied ethnicity.
Topical glucocorticoids are indicated for oral and genital ulcers. Colchicine is the drug of choice for prevention of recurrent mucocutaneous lesions (especially erythema nodosum or genital ulcers) and acute arthritis. Azathioprine, interferon alfa, or tumor necrosis factor (TNF)-α inhibitors should be considered in recurrent and chronic arthritis. Management of uveitis requires close collaboration with an ophthalmologist. Glucocorticoids and other immunosuppressives are recommended to prevent relapse of deep venous thromboses. Glucocorticoids should be considered during acute exacerbations of nonsurgical abdominal pain, together with 5-ASA or azathioprine. Acute attacks of the central nervous system are treated with high-dose glucocorticoids and azathioprine. For more severe aspects of Behçet syndrome, treatment resistance warrants consideration of infliximab or adalimumab, or, in some cases, cyclophosphamide.
Relapsing Polychondritis
Relapsing polychondritis (RP) is a rare autoimmune disease characterized by inflammation and damage of cartilaginous tissues. Anti-type II collagen antibodies have been identified, but a role in pathogenesis has not been established. RP can be primary or associated with other autoimmune diseases, particularly ANCA-associated vasculitis, Behçet syndrome, and antiphospholipid syndrome. Onset usually occurs between the ages of 40 and 50 years.
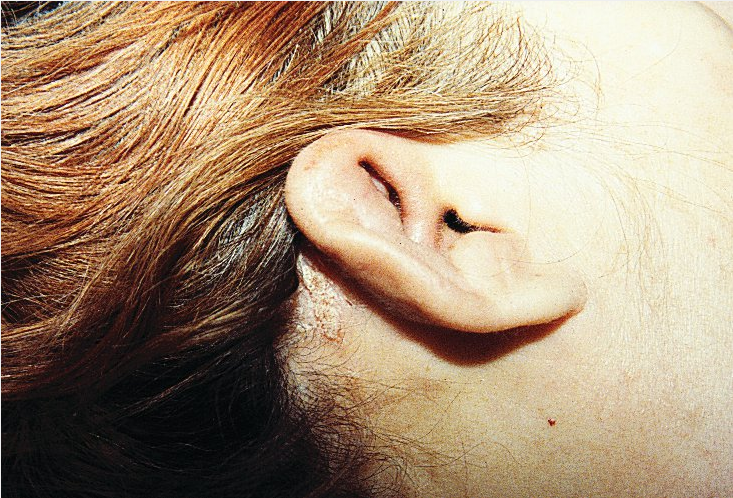
Tissues affected include the cartilaginous portions of the external and middle ear (90%), nose (60%), tracheobronchial tree (50%), and joints (65%). Inflammation of noncartilaginous connective tissue also occurs, particularly uveitis and keratitis (65%). Cochlear and/or vestibular dysfunction may rarely occur (10%). Auricular involvement affects the helix but spares the earlobes. Nasal chondritis can result in collapse of the nasal bridge (saddle nose deformity), which can also be seen in trauma, granulomatosis with polyangiitis, cocaine use, congenital syphilis, and leprosy. Airway stenosis from tracheal ring involvement and aortitis/large-vessel vasculitis may occur and be life-threatening. RP is diagnosed by its typical clinical manifestations.
Elevated inflammatory markers, anemia, antinuclear antibodies, and other autoantibodies are often observed but are nonspecific. Cartilage biopsy, which is necessary only with atypical presentations in which there is substantial diagnostic uncertainty, typically demonstrates inflammatory cell infiltration of the perichondrium. CT can demonstrate bronchial thickening, strictures, malacia, and air trapping.
Therapy is guided by organ involvement and disease severity. Mild disease may respond to NSAIDs, colchicine, or dapsone. Glucocorticoids and disease-modifying antirheumatic drugs (DMARDs) such as cyclosporine, azathioprine, or methotrexate are used for more severe disease. Biologic drugs such as TNF-α inhibitors have been reported as effective but not on a consistent basis. Surgery (stenting, dilation, extirpation, reconstruction) for airway lesions may be necessary.
Adult-Onset Still Disease
Adult-onset Still disease (AOSD) is a rare disorder that affects multiple organ systems. Symptoms and signs include daily high spiking fever, evanescent salmon-colored rash on the trunk and extremities, arthritis, lymphadenopathy, and leukocytosis. It is most commonly diagnosed in young adults (median age, 36 years) but has been described in older age groups. Some reports indicate a predilection for women. The cause is unknown, but AOSD appears to be a disease of autoinflammation rather than autoimmunity.
See Table 43 for diagnostic criteria. Diagnosis is clinical, and other infectious, neoplastic, and rheumatologic diseases must be excluded. Rare complications include hemophagocytic lymphohistiocytosis, myocarditis, shock, multiple organ failure, disseminated intravascular coagulation, thrombotic microangiopathy, and fulminant hepatitis.

Treatment includes NSAIDs, glucocorticoids, DMARDs such as methotrexate, and IL-1 inhibitors. TNF-α and IL-6 inhibitors may be beneficial, but more studies are needed to establish their efficacy. Most patients respond to therapy, and prognosis of AOSD with treatment is good.
Autoinflammatory Diseases
Autoinflammatory diseases (periodic fever syndromes) are a category of rare monogenic conditions characterized by episodic and/or persistent inflammation in the absence of antigenically driven autoimmunity. Pathogenesis involves dysregulation of the innate immune system. The innate immune system represents a process separate and distinct from the antigen-based immune response modulated by lymphocytes. Rather than adaptively responding to antigens, the innate immune system responds to stimuli that it “innately knows” to mandate a response. Examples of such stimuli are “pathogen-associated molecular patterns” such as bacterial cell wall proteins and “damage-associated molecular patterns” or markers of cellular damage such as released uric acid. This process is largely regulated by “inflammasomes,” large protein structures that recognize these stimuli. Autoinflammatory diseases involve inherited or spontaneous mutation of genes that encode for inflammasomes, including components of the IL-1β–activating inflammasome and the TNF-α receptor pathway.
See Table 44 for the features and treatment of select autoinflammatory diseases.
Sarcoidosis
In patients with sarcoidosis, a periarthritis involving multiple joints may occur, typically affecting the ankles but also potentially the knees, wrists, and small joints of the hands and feet. Erosive damage, deformity, and dactylitis may develop. Bone involvement has been reported in 1% to 15% of patients; it most commonly accompanies multiorgan disease and is associated with a chronic disease course and poorer prognosis. Typical radiographic findings include cystic or sclerotic lesions and a lacy pattern of multiple lesions (Figure 31).
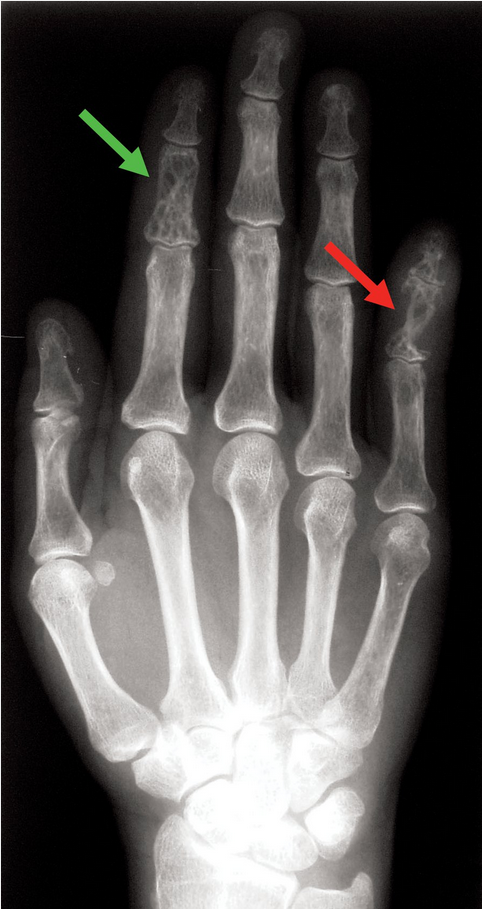 Hand radiograph showing the typical cystic lesions (red arrow) and a lacy pattern (green arrow) characteristic of sarcoidosis.
Hand radiograph showing the typical cystic lesions (red arrow) and a lacy pattern (green arrow) characteristic of sarcoidosis.
Sarcoid myopathy is histologically present in 25% to 75% of patients but is symptomatic in only 0.5% to 5%. The most common clinical presentation is chronic proximal weakness with development of atrophy or contractures. Biopsy may show inflammatory muscle disease or destruction of muscle by granuloma formation. Glucocorticoids, DMARDs, and TNF-α inhibitors may be used to treat arthritis, bone disease, and myopathy.
Rarely, sarcoidosis can cause Heerfordt syndrome (uveoparotid fever), a combination of fever, uveitis, and parotitis with or without cranial nerve VII palsy. This can lead to sicca symptoms and can mimic Sjögren syndrome.
Löfgren syndrome is a sarcoidosis triad of bilateral hilar lymphadenopathy, erythema nodosum, and migratory polyarthralgia. Fever is frequently present. When all three parts of the triad are present, there is a 95% diagnostic specificity for sarcoidosis, obviating the need for biopsy. Löfgren syndrome has a good prognosis and usually remits in 2 to 16 weeks. The goal of treatment is symptom reduction; NSAIDs are usually adequate, but low-dose glucocorticoids, colchicine, and hydroxychloroquine can all be helpful.
Genetic Diseases of Connective Tissue
Mutations in genes encoding for collagen, fibrillin, and other components of connective tissue can lead to poorly functional skin and integument. Common clinical findings include skin and joint laxity, skeletal fractures, and cardiovascular complications related to involvement of the aorta and cardiac valves. The three most common syndromes are Ehlers-Danlos syndrome (EDS), Marfan syndrome (MFS), and osteogenesis imperfecta (OI). See Table 45 for a summary of clinical findings and management of these syndromes.
Vascular monitoring is necessary in MFS and several types of EDS. Patients with MFS should undergo echocardiography to assess the aortic root and ascending aorta at the time of diagnosis and at 6 months to ensure stability. Follow-up studies should be completed at least annually; MRI and/or CT are often used to further delineate risk. The best means of vascular surveillance in vascular EDS is unclear, and all patients should undergo expert consultation. See MKSAP 18 Cardiovascular Medicine for more information.
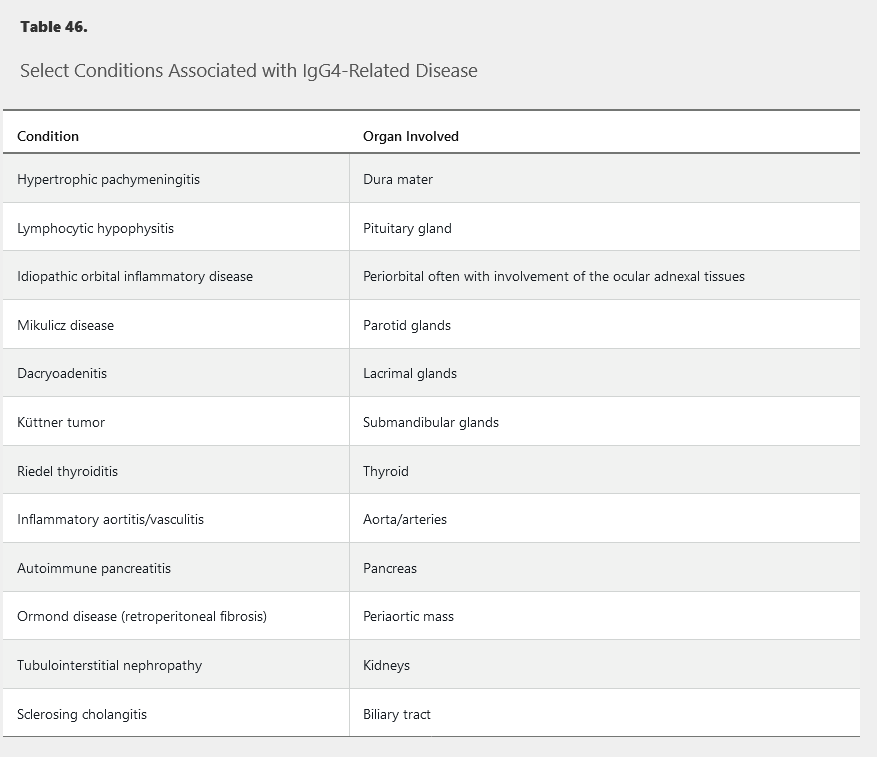
IgG4-Related Disease
Several previously ill-defined illnesses have been brought under the banner of IgG4-related disease (IgG4-RD) (Table 46). Most of these conditions are characterized by infiltration and tumefaction of the affected tissue with resultant organ enlargement, fibrosis, and dysfunction. Patients commonly present with a sentinel organ enlargement, but careful evaluation often reveals more extensive disease.
Currently, IgG4 is hypothesized not to be pathologic, but instead to act as a blocking antibody in patients with atopic disease, preventing allergens from interacting with IgE on mast cell surfaces. Consistent with this model, IgG4 elevations accompany loss of clinical responses to allergens in atopic individuals.
Most patients with IgG4-RD are men over the age of 50 years; history of atopic disease is common. Clinical signs include painless enlargement of lymph nodes or the thyroid, parotid, or submandibular glands; proptosis with orbital pseudo-tumor; back or chest pain from aortic involvement; and abdominal pain from pancreatic or biliary tree disease. Systemic symptoms such as fever or weight loss are uncommon.
IgG4-RD can affect vascular structures directly, including the carotid, pulmonary, coronary, and iliac arteries as well as the aorta, in particular the thoracic aorta. The affected vessels can develop aneurysms, dissection, and stenosis. A periarteritis primarily affecting the infrarenal aorta in conjunction with retroperitoneal fibrosis may also develop.
Diagnosis is by tissue biopsy, which demonstrates a dense lymphoplasmacytic infiltrate, CD4-positive T cells and plasma cells in germinal centers, IgG4-staining plasma cells, storiform fibrosis, obliterative phlebitis or arteritis, and tissue eosinophilia.
IgG4 levels are elevated in the serum in 70% to 80% of patients; therefore, a normal serum level does not rule out the disease. A ratio of the IgG4 level to total IgG of more than 8% is suggestive of the diagnosis. PET scan can identify involvement that is not clinically apparent and can be used to document response.
Initial treatment is prednisone, 0.5-0.6 mg/kg/d, for 2 to 4 weeks with a slow taper over 3 to 6 months; some experts advocate subsequent low-dose (5 mg/d) therapy for several years. Rituximab has shown significant benefit and appears to affect IgG4 production more than other IgG subtypes. Methotrexate may be a useful agent to maintain remission in patients treated with prednisone or rituximab. Azathioprine and mycophenolate mofetil have also been used as glucocorticoid-sparing agents. Treatment response may be influenced by the amount of fibrosis present prior to therapy initiation.