Neuromuscular Disorders
- related: Neurology
- tags: #neurology
Overview
Peripheral and central nervous system disorders can be differentiated on the basis of history and neurologic examination findings (Table 44). Neuromuscular disorders include myopathies, neuromuscular junction disorders, peripheral neuropathies, plexopathies, radiculopathies, and motor neuron diseases (Table 45 and Table 46).
Peripheral Neuropathies
Classification, Findings, and Diagnosis
Symptoms of peripheral neuropathy include negative (sensory loss) or positive (paresthesia, dysesthesia, and pain) sensory symptoms, weakness, or autonomic dysfunction. Neuropathies are classified on the basis of distribution of sensorimotor deficits (symmetry, distal versus proximal, focal versus generalized), pathology (demyelinating versus axonal), family history, and autonomic involvement (Figure 28).
For all patients with a suspected peripheral neuropathy, a complete blood count, an erythrocyte sedimentation rate determination, serum protein electrophoresis with immune fixation, thyroid function tests, hemoglobin A1c/fasting plasma glucose determination, and vitamin B12 measurement are necessary. In specific clinical settings, additional tests, including CSF analysis, genetic testing, and other specialized laboratory testing, can help clarify the diagnosis (Table 47).
Electromyography (EMG), including both nerve conduction studies and needle electrode examination, can differentiate neuropathy from other neuromuscular disorders. In neuropathy, motor unit potentials are high amplitude and prolonged, whereas in myopathy, these potentials are low amplitude and brief. If neuropathy is present, EMG also can determine whether it is demyelinating or axonal. In demyelinating neuropathies, velocity of nerve conduction decreases, but in axonopathy, velocity remains unchanged. The presence of weakness, asymmetry, proximal findings, rapid course, or atypical features should prompt EMG testing. In patients with a slowly progressive distal symmetric sensory neuropathy with negative family history, however, EMG may not be required.
Nerve biopsy should only be considered in rare settings in which concern for a vasculitic, infectious, or infiltrative neuropathy exists. Autonomic tests, including quantitative sudomotor axon reflex and tilt table tests, can help confirm autonomic neuropathy but are rarely performed in practice.
Mononeuropathies
Mononeuropathies involve a single nerve and can cause both motor and sensory symptoms within the territory of the affected nerve. Sequential involvement of multiple nerves may be seen in mononeuropathy multiplex, a condition most often associated with pain and caused by vasculitis. Plexopathies originate at the level of the brachial or lumbosacral plexus and involve multiple sensory and motor nerves simultaneously.
Carpal Tunnel Syndrome
Carpal tunnel syndrome results from focal compression of the median nerve at the wrist. Most often, paresthesia and pain are experienced in the first three digits but may radiate to the entire hand or proximally. With milder symptoms, diagnosis of carpal tunnel syndrome can be made clinically, and treatment is supportive, including neutral positioning, wrist splints, and pain control. The presence of weakness, thenar atrophy, refractory pain, or active denervation evident on EMG should prompt surgical decompression. See MKSAP 18 General Internal Medicine for further information about carpal tunnel syndrome.
Brachial and Lumbosacral Plexopathies
Common causes of brachial and lumbosacral plexopathy include diabetes, malignancy, radiation, and trauma. Imaging is mandatory to rule out structural lesions and malignancy. Idiopathic brachial plexitis (also known as neuralgic amyotrophy) presents with subacute severe scapular pain followed by pronounced weakness and atrophy in shoulder-girdle and upper extremity muscles. It is likely immune mediated, and a triggering event, such as infection or surgery, usually precedes this presentation, although the infection or surgery is not in the region of the brachial plexus. EMG can confirm the diagnosis, and imaging can rule out alternative causes. Treatment is supportive. Gradual recovery (90% by 3 years) is common. Idiopathic lumbosacral radiculoplexus neuropathy is immune mediated and very similar in presentation to diabetic amyotrophy. (See Neuropathies of Diabetes Mellitus and Impaired Glucose Tolerance.)
Polyneuropathies
Polyneuropathies arise from various disorders causing injury to peripheral nerves (see Table 47). Polyneuropathies may affect either large nerve fibers (and lead to impaired proprioception, hyporeflexia, and weakness) or small nerve fibers (and cause pain, dysesthesia, and dysautonomia). Additionally, they may be primarily axonal or demyelinating. If axonal, the distal axonal segments are affected first, which leads to a length-dependent stocking-glove pattern of involvement. A sensory-predominant distal symmetric neuropathy is typically seen with metabolic, toxic, and systemic neuropathies. Demyelinating neuropathies, which result from injury to the myelin sheath of the peripheral nerve, can cause non–length-dependent weakness and sensory loss.
Neuropathies of Diabetes Mellitus and Impaired Glucose Tolerance
Diabetes mellitus causes various types of neuropathy (Table 48). The most common presentation is symmetric distal neuropathy involving small and large sensory fibers and, to a lesser extent, distal motor nerves. Clinical examination may reveal length-dependent loss of light touch and vibration sensation, loss of the ankle reflex, and—in advanced disease—distal weakness. Pain and paresthesia may predominate, especially when small fibers are involved. Patients with a pure small-fiber variant of symmetric distal neuropathy may have normal distal sensory and reflex findings on examination and normal results on EMG at presentation; this pattern also can be seen at various stages of diabetes and even in patients with impaired glucose tolerance. Between 11% and 25% of patients with glucose intolerance have evidence of small-fiber neuropathy on specialized testing, but only a minority (5%-10%) are symptomatic.
Autonomic diabetic neuropathy can cause orthostatic hypotension, erectile dysfunction, abnormal hidrosis, and gastroparesis. Dysautonomia also can mask symptoms of hypoglycemia and may predispose patients to silent myocardial infarction. Diabetic mononeuropathy can involve the cranial nerves (especially III, VI, and VII) and truncal nerve roots (with a dermatomal pattern of pain and paresthesia in the chest or abdomen) and predispose patients to entrapment neuropathies, such as carpal tunnel syndrome.
Diabetic amyotrophy, also known as proximal lumbosacral radiculoneuropathy, is associated with subacute painful involvement of the lumbosacral plexus followed by resolution of pain and onset of marked asymmetric weakness with atrophy and weight loss. The differential diagnosis of diabetic amyotrophy includes polyradiculopathy, retroperitoneal hematoma, neoplasm, carcinomatous meningitis, and inflammatory neuropathy. Spontaneous recovery over 1 to 3 years is typical but may be incomplete.
Tight glycemic control, exercise, and management of dyslipidemia, obesity, and metabolic syndrome can slow the progression and improve the symptoms of diabetic polyneuropathy. Management of diabetic amyotrophy consists of supportive measures, physical therapy, and pain control but does not involve immunosuppression. See MKSAP 18 Endocrinology and Metabolism for further information about diabetic neuropathy, including neuropathic pain treatment.
Hereditary Neuropathies
Charcot-Marie-Tooth disease consists of more than 70 genetic disorders in which peripheral neuropathy is the only or main clinical manifestation. The most common inherited neuropathy, Charcot-Marie-Tooth disease type 1 is an autosomal dominant demyelinating neuropathy presenting with early onset, slowly progressive distal weakness, areflexia, and sensory loss without pain or paresthesia. Foot deformities, such as hammer toes, high arches, and distal leg atrophy (stork legs) are common (Figure 29). EMG typically reveals a uniform slowing of nerve conduction velocities, and genetic testing is confirmatory.
Inflammatory Polyradiculoneuropathies
Guillain-Barré Syndrome
Guillain-Barré syndrome (GBS) is an acute autoimmune demyelinating polyradiculoneuropathy that presents with rapidly progressive flaccid weakness. Onset is often preceded by a respiratory or gastrointestinal infection, triggering a T-cell–mediated autoimmune attack against peripheral nerve and root myelin. Weakness is ascending, starting in the lower extremities, spreading to the upper limbs and bulbar and respiratory muscles, and reaching its nadir in less than 4 weeks. The weakness may occur first in the proximal muscles. Paresthesia and early low back pain are common. Dysautonomia can be severe and predispose patients to labile blood pressure and arrhythmias. Progressive respiratory and bulbar weakness may lead to rapid respiratory failure. On examination, diffuse areflexia is a key finding; there is not marked sensory loss, even in the setting of paresthesia. GBS variants include demyelinating polyradiculoneuropathy involving only or sparing the legs and the Miller-Fisher variant, which presents with ataxia, cranial neuropathies, and antibodies to GQ1b ganglioside protein (a highly sensitive finding).
The differential diagnosis of GBS includes botulism (descending weakness), myasthenic crisis (without pain and paresthesia), acute myelopathies (hyperreflexia and upper motor neuron signs), West Nile virus (asymmetric paralysis), carcinomatous and sarcoid meningitis (slower course), Lyme disease, HIV seroconversion, porphyria, and tick paralysis.
CSF analysis shows a pattern of a highly elevated protein level with a normal or mildly elevated leukocyte count (albuminocytologic dissociation) in 90% of patients. EMG is the confirmatory test and shows a predominantly demyelinating pattern; sensitivity is initially low but 90% by week 5. Initiation of treatment should not await EMG confirmation; a negative early study should be repeated later if the diagnosis remains suspected. Additional studies, such as spinal MRI and serologic testing for Lyme disease, HIV, and myasthenia antibodies, may be considered in appropriate clinical settings.
Therapy includes supportive management and immunotherapy. All patients with GBS should be hospitalized for close respiratory and cardiovascular monitoring. Decisions about intubation should be guided by measurement of forced vital capacity and negative inspiratory force and be made before emergence of hypoxemia. Both plasmapheresis and intravenous immune globulin (IVIG) have equal efficacy in shortening the time to recovery and the duration of ventilation. Serial treatment with IVIG after plasma exchange is not superior to either treatment alone and should not be considered unless symptoms worsen after initial improvement or stabilization. Glucocorticoids are contraindicated in GBS and may worsen outcome. Prognosis is favorable, with 80% of patients resuming ambulation by 6 months after onset. Relapse occurs in 6% of patients with GBS and requires repeated treatment.
Chronic Inflammatory Demyelinating Polyradiculoneuropathy
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a potentially treatable autoimmune neuropathy. CIDP often presents with a progressive or relapsing symmetric proximal and distal weakness and sensory symptoms with diffuse areflexia. Its temporal course is often subacute with progression after 8 weeks of onset, which helps to differentiate CIDP from acute inflammatory demyelinating polyneuropathy (AIDP aka gbs guillain barre syndrome). Atypical forms include multifocal asymmetric and distal symmetric variants, but respiratory failure is rare. CIDP may be isolated or occur in the setting of several other systemic conditions, including diabetes mellitus, lymphoma, and HIV. CSF findings are similar to those of GBS and may show albuminocytologic dissociation, and a demyelinating pattern on EMG is the key to diagnosis. Nerve biopsy is often unnecessary but, in complex presentations, can differentiate CIDP from vasculitis or amyloidosis. First-line treatments include glucocorticoids, periodic IVIG, or plasma exchange, which all have similar efficacy. Treatment is typically continued for at least 6 months. Half of patients achieve remission, but the other half relapse and require resumption of immunotherapy. Second-line therapies, including azathioprine, mycophenolate mofetil, cyclosporine, or cyclophosphamide, are often used off-label to treat refractory disease.
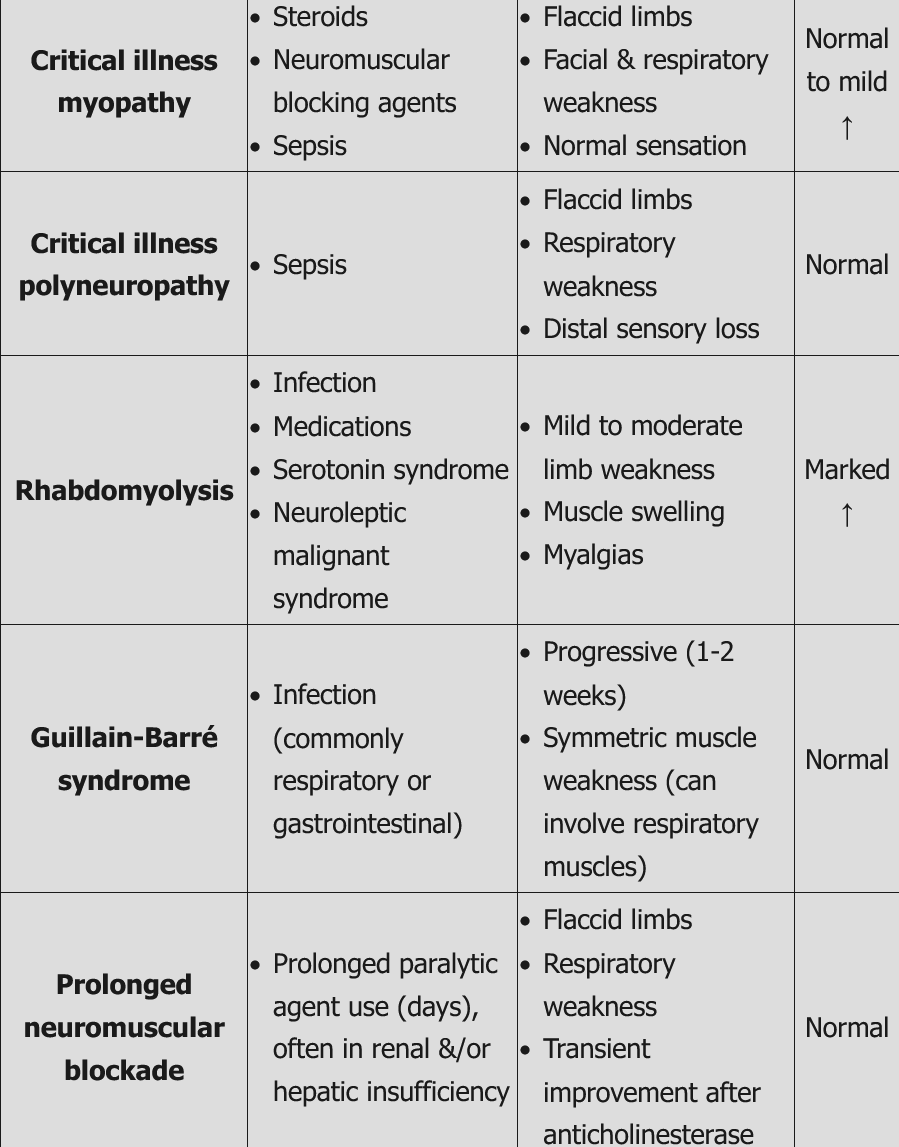
Critical Illness Neuropathy
Prolonged intensive care treatment, particularly in association with sepsis and multiorgan failure, can lead to diffuse weakness, an inability to wean patients from ventilators, and prolonged posthospitalization weakness. Weakness may be secondary to critical illness myopathy, critical illness axonal polyneuropathy, or a combination of the two. Critical illness myopathy is more common, less severe, and potentially caused by channelopathy (a disease involving dysfunction of a cellular ion channel) triggered by sepsis and systemic disease. Critical illness neuropathy, on the other hand, is associated with microcirculatory axonal damage and a more protracted recovery. EMG can differentiate between the two, but often components of both are simultaneously present; in rare cases, nerve and muscle biopsy may be necessary. Differential diagnosis includes GBS, myasthenia gravis, vasculitis, and central weakness. No definitive treatment is available, but appropriate glycemic control, early mobilization and therapy, and minimizing glucocorticoid use improve prognosis. One third of patients with critical illness neuropathy die during the acute phase of the disease, with the remainder experiencing a full or partial slow recovery within months to years.
This patient’s presentation is consistent with critical illness myopathy (CIM), which is commonly seen in ICU patients with asthma or COPD treated with steroids. Risk factors for CIM include systemic corticosteroids (strongest risk factor), neuromuscular blocking agents, and sepsis. Patients usually present several days after IV steroid or paralytic agent administration with flaccid quadriparesis (proximal > distal muscles) and/or difficulty weaning from mechanical ventilation. Facial muscle weakness may also occur. Sensation is usually normal, but reflexes may be decreased. Critical illness myopathy is more common than critical illness polyneuropathy (CIP), but patients sometimes have a combination of the two.
Diagnosis is suspected clinically and can be confirmed by EMG/NCS. Serum creatine kinase levels can be normal or mildly elevated. Muscle biopsy can also show myopathy with loss of myosin. Treatment of CIM involves stopping or reducing steroids as soon as possible, aggressive management of underlying medical conditions, and occupational and physical rehabilitation. Prognosis is generally good with reversal of symptoms over weeks to months, but recovery may require prolonged hospitalization.
Paraproteinemic Neuropathy
Paraproteinemic neuropathy frequently presents as a symmetric distal sensory neuropathy, but sensorimotor, multifocal motor, or cranial nerve variants also are possible. Therefore, all patients with peripheral neuropathy should be screened for paraproteinemia. Neuropathies associated with monoclonal proteins occur in monoclonal gammopathy of undetermined significance, multiple myeloma, Waldenström macroglobulinemia, amyloidosis, and other hematologic malignancies. (See MKSAP 18 Hematology and Oncology.) Diagnosis depends on the detection of monoclonal proteins, and treatment is based on the underlying condition. A CIDP-like severe polyneuropathy may occur in paraproteinemic neuropathy in association with organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes (POEMS syndrome); this syndrome always is associated with λ paraproteinemia. Treatment is similar to that of multiple myeloma and may reverse neuropathic symptoms. Another neuropathic disorder associated with paraproteins is distal acquired demyelinating symmetric neuropathy. Patients with this condition generally are responsive to immunosuppressive therapy (glucocorticoids, IVIG, and rituximab), except for those with antibodies to myelin-associated glycoprotein; their disease is medication refractory.
Autonomic Neuropathy
Autonomic neuropathies manifest as diffuse or focal impairment of cholinergic or sympathetic autonomic systems. Whereas diabetes, amyloidosis, and HIV are common disorders underlying secondary autonomic neuropathy, primary autoimmune ganglionopathy is an important and potentially treatable cause of autonomic neuropathy that is often associated with antibodies against ganglionic nicotinic acetylcholine receptors. The presentation may range from acute pandysautonomia to chronic focal autonomic dysfunction. Antibody testing and skin biopsy help with diagnosis. Response to IVIG, plasmapheresis, or immunosuppression is common.
Amyloid neuropathy is caused by extracellular deposition of amyloid protein in peripheral nerves and other tissues. Amyloidosis starts with painful small-fiber neuropathy and progressively causes major autonomic impairment and weakness and multiorgan involvement. Primary amyloidosis is associated with monoclonal proteins, whereas familial amyloidosis is secondary to transthyretin gene mutation. See MKSAP 18 Hematology and Oncology.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of motor neurons that causes progressive weakness, atrophy, and (eventually) death. This motor neuron disease often begins as isolated extremity or bulbar weakness (dysphagia, dysarthria) but relentlessly spreads to other regions. Upper (hyperreflexia, spasticity, and extensor plantar response) and lower (atrophy and fasciculation) motor neuron signs and the absence of sensory deficits are characteristic. Frontotemporal dementia occurs in 50% of affected patients, but oculomotor palsy, incontinence, and tremor are atypical. Diagnosis is based on clinical evidence of both upper and lower motor neuron signs on examination and EMG evidence of lower motor neuron signs in at least two (probable ALS) or more (definite ALS) regions. Alternative diagnoses also must be excluded by brain and cervical spinal imaging and laboratory testing; cervical cord compression (causing lower motor neuron signs at the level of compression and some upper signs below that level), vitamin B12 and copper deficiencies, Lyme disease, hyperparathyroidism, and thyrotoxicosis must be excluded. Multifocal motor neuropathy is a treatable ALS mimic characterized by severe weakness with minimal atrophy, absence of upper motor neuron signs, and EMG evidence of motor conduction block. Benign fasciculation syndrome causes widespread fasciculation without weakness or upper motor neuron signs and should not be confused with ALS.
Treatment of ALS is supportive and should involve multidisciplinary care. Riluzole, a glutamate release blocker, is an FDA-approved treatment for ALS and may increase survival by 3 months. Edaravone, an intravenous free radical scavenger, recently received FDA approval on the basis of evidence showing that it slowed functional decline in ALS within a 6-month period. Whether this agent can slow the course of ALS over a longer period or can reduce mortality remains under investigation. Noninvasive ventilation, nutritional support (including percutaneous endoscopic gastrostomy), and treatment of pseudobulbar affect by dextromethorphan-quinidine also have shown some benefit. Prognosis and goals of care should be discussed with the patient early in the disease course to avoid unnecessary diagnostic and therapeutic measures.
Neuromuscular Junction Disorders
Myasthenia Gravis
Myasthenia gravis (MG) is an autoimmune disease of the postsynaptic neuromuscular junction associated with antibodies to the postsynaptic acetylcholine receptors. Onset most commonly occurs in the third decade of life in women and after age 50 years in men. Ptosis and diplopia are the first manifestations in two thirds of patients, although only half of these patients develop generalized myasthenia, typically within 2 years. Early bulbar and cervical involvement is seen in 10% of patients. Fluctuating painless weakness without sensory loss is typical. Weakness may be missed on clinical examination unless fatigability (fatigable diplopia) is assessed by sustained or repeated activation of muscles. Spared pupillary response helps distinguish MG from Horner syndrome, and involvement of the extraocular muscles (oculomotor, trochlear, and abducens nerves [CNs III, IV, and VI]) and palate (glossopharyngeal and vagus nerves [CNs IX and X]) helps distinguish MG from Bell palsy (facial nerve [CN VII]). The presence of respiratory symptoms should prompt close monitoring of respiratory parameters because of the risk of rapid respiratory failure (myasthenic crisis). This crisis can occur as part of the natural history of bulbar or generalized myasthenia or be triggered by external factors, including infection, surgery, or certain medications (especially aminoglycosides, quinolones, magnesium, beta blockers, and hydroxychloroquine).
Diagnosis of MG is based on clinical, serologic, and EMG findings. Disease-specific antibodies are found in 90% of patients; of these, 85% have typical MG with acetylcholine receptor antibodies, but 5% have anti–muscle-specific kinase (MuSK) antibodies. MuSK-positive myasthenia is more likely to cause focal or severe bulbar, cervical, or respiratory weakness. The characteristic EMG finding of MG is a decremental response to repetitive stimulation. All patients with MG should undergo chest CT to screen for thymoma, a tumor associated with the disease. The edrophonium and ice pack tests are no longer recommended as first-line testing due to high false-positive results.
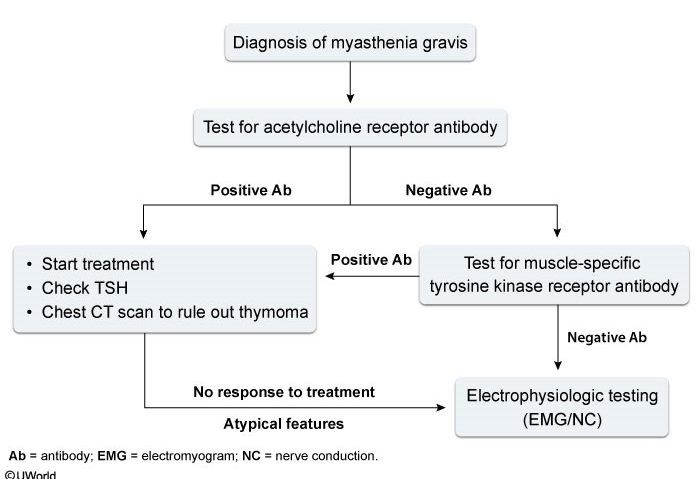
Symptomatic treatment of ocular and mild generalized myasthenia usually starts with the cholinesterase inhibitor pyridostigmine. In those with more advanced disease, immunosuppressive therapy is required. Oral glucocorticoids often are used as first-line treatment but can cause transient exacerbation at high doses and should be titrated upward slowly in patients with mild to moderate weakness. In the presence of prominent bulbar or generalized weakness and in myasthenic crisis, treatment with IVIG or plasmapheresis should precede initiation of glucocorticoids. The immunosuppressant agents azathioprine, mycophenolate mofetil, and cyclosporine are effective long-term maintenance therapies. However, these drugs have a delayed onset of action, and bridging therapy with concomitant glucocorticoids, IVIG, or plasmapheresis is usually required.
TSH should be checked in all patients due to MG's high association with autoimmune thyroid abnormality. In patients with high suspicion of other autoimmune disorders, testing (eg, antinuclear antibody panel) is indicated as appropriate.
Thymectomy should be performed in all patients with thymoma. Benefits of thymectomy in patients without thymoma are not firmly established, but a recent randomized controlled trial confirmed that thymectomy improves clinical outcome and reduces immunosuppression requirements in patients with generalized MG who are younger than 65 years and within 3 years of diagnosis.
MuSK-positive myasthenia responds well to plasmapheresis and glucocorticoids but requires aggressive maintenance immunosuppression. Prolonged remission has been reported with rituximab (off-label use); thymectomy and pyridostigmine are not helpful.
Lambert-Eaton Myasthenic Syndrome
Lambert-Eaton myasthenic syndrome is an autoimmune disorder of the presynaptic neuromuscular junction associated with antibodies against the voltage-gated calcium channel. This disorder presents similarly to MG, except that weakness improves with exercise, and hyporeflexia and dysautonomia are present. Diagnosis is confirmed by detection of serum anti–voltage-gated calcium channel antibodies (90%) and the EMG finding of augmented motor response to rapid repetitive stimulation. Malignancy, especially small cell lung cancer, is found in half of patients. Treatment consists of treating any underlying malignancy or, in nonparaneoplastic disease, immunosuppression, IVIG, or plasmapheresis.
Myopathies
Overview
Classification of myopathies is based on clinical and pathologic characteristics and acquired versus inherited causes (Table 49). Most myopathies involve symmetric weakness of the proximal limb muscles. A normal sensory and reflex examination can differentiate myopathy from neuropathy. Additional features that can indicate a specific diagnosis include the clinical course; the presence of pain and stiffness; atypical distribution in ocular, bulbar, or distal limb muscles; and episodic symptoms (Table 50).
Diagnosis is based on systematic clinical assessment, muscle-related serum markers (creatine kinase [CK], aldolase), EMG findings, muscle biopsy, and, in certain cases, genetic testing. The serum CK level is elevated in many forms of myopathy and can be followed to monitor disease activity and response to treatment in inflammatory myopathies. Mild elevation of the serum CK level (<5 times normal) is not specific to myopathies and also can be seen in ALS, CIDP, muscle trauma, and persistent elevation of the serum CK level without weakness (benign hyperCKemia). EMG can confirm the presence of changes associated with myopathy (low amplitude, short duration, polyphasic shape of motor unit potentials), determine distribution of involved muscles, and rule out neuropathy or a neuromuscular junction disorder. The serum CK level should be checked before EMG to prevent procedure-related false positive results. Muscle biopsy is the most helpful test to confirm the diagnosis of myopathy, but in some hereditary myopathies, genetic testing can provide the confirmation without need for biopsy.
Inflammatory Myopathy
Polymyositis, dermatomyositis, immune-mediated necrotizing myopathy, and inclusion body myositis are idiopathic inflammatory myopathies. Polymyositis and dermatomyositis present with acute or subacute proximal muscle weakness. Diagnosis is based on an elevated serum CK level, EMG findings, and muscle biopsy findings. Immune-mediated necrotizing myopathy typically presents with progressive proximal weakness and an elevated serum CK level; diagnosis is based on muscle biopsy findings of necrotic fibers with limited inflammation. Onset can be triggered by statins, but weakness continues to progress after removal of the drug. In this setting, the presence of the disease-specific serum antibody to hydroxymethylglutaryl coenzyme A reductase can support the diagnosis. Treatment with immunosuppression reverses the myopathy.
Inclusion body myositis has a slowly progressive course and causes early weakness and atrophy of distal upper extremity flexors, quadriceps, and bulbar muscles. Muscle biopsy reveals inflammation and characteristic inclusion bodies. See MKSAP 18 Rheumatology for further information on inflammatory myopathy.
Endocrine-Related Myopathy
Hypothyroid myopathy can cause diffuse myalgia, proximal weakness, and myoedema (muscle mounding after percussion). Hyperthyroidism also can cause myopathy in association with fasciculation, ophthalmoplegia and hyperreflexia.
Glucocorticoid-Induced Myopathy
Exposure to chronic high-dose exogenous glucocorticoids can cause myopathy of unclear mechanism. This disorder is associated with proximal weakness and myalgia but normal CK levels and (mostly) normal EMG findings. Dexamethasone is more likely than prednisone or hydrocortisone to cause this type of myopathy. In patients treated with glucocorticoids for inflammatory myopathies, persistence of weakness after normalization of the CK level may indicate glucocorticoid-induced myopathy, and a trial of glucocorticoid tapering may be warranted.
Toxic Myopathy
Toxic myopathy can be triggered by statins and other drugs (see Table 49). Statins can cause acute toxic myopathy associated with rhabdomyolysis. The lipophilic statins metabolized by the cytochrome P450 3A4 isozyme system (simvastatin, atorvastatin, and lovastatin) have a higher propensity to cause myopathy than the hydrophilic agents pravastatin and rosuvastatin. The risk of myopathy increases with higher dosages, the addition of fenofibrate or gemfibrozil, and the addition of cytochrome P450 3A4 isozyme inhibitors. Serum CK levels can be mildly or highly elevated in toxic myopathy.
Inherited Myopathies
Inherited myopathies are listed in Table 49. Many of these disorders develop early in life, but some present in adulthood. Myotonic dystrophies are systemic diseases associated with myotonia, an impairment of muscle relaxation causing stiffness and a delayed hand-grip release. Myotonic dystrophy type 1 causes distal weakness and is associated with cataracts, frontal balding, cardiac and endocrine disease, and mild cognitive impairment. Progression of weakness is slow, but diagnosis should prompt close monitoring for cardiac and pulmonary disease that can cause premature mortality.
Mitochondrial myopathy presents with significant variability and can cause fatigue, myalgia, ophthalmoplegia, and various extramuscular manifestations. Mitochondrial myopathies should be suspected in the presence of a fluctuating course, multiorgan involvement, and maternal transmission.
Adult-onset metabolic myopathies may present with isolated exercise-induced weakness, cramps, and myoglobinuria and include many deficiencies of key metabolic pathways, such as glycogen storage and fatty acid oxidation; the most common types are carnitine palmitoyltransferase II deficiency and McArdle disease.
Acid maltase deficiency (Pompe disease) has an adult-onset form associated with proximal and respiratory muscle weakness. Diagnosis is based on assessment of α-glucosidase activity and genetic testing. Alglucosidase alfa is FDA approved to prevent progression of weakness.