FSGS
- related: Nephrology
- tags: #nephrology
Epidemiology and Pathophysiology
Focal segmental glomerulosclerosis (FSGS) is the most common form of the nephrotic syndrome in Black patients and, in certain parts of the world, has replaced membranous glomerulopathy as the leading cause of the nephrotic syndrome in White patients. The prevalence of FSGS in the United States is more than 20,000 patients, with a yearly incidence of approximately 5000 newly diagnosed cases per year. In the United States, FSGS currently accounts for up to 40% of primary nephrotic syndromes in adults. Worldwide, the incidence of FSGS has been estimated at nearly 1 in 100,000 people per year.
FSGS stems from abnormalities in the podocyte. Podocyte detachment and death lead to the segmental sclerosis that is the hallmark histopathology of FSGS. The podocyte injury in FSGS, in turn, can stem from immunologic, genetic, and/or hyperfiltration causes. Immunologic injury is considered the main pathogenic mechanism behind primary forms of FSGS, with leukocytes producing a soluble circulating factor that directly targets podocytes. This mechanism is supported by cases of FSGS recurring almost immediately after kidney transplantation and responding briskly to plasmapheresis, although to date definitive identification of such a circulating factor has yet to occur. Identified genetic causes of FSGS are chiefly mutations in podocyte-specific proteins (for example, nephrin, podocin, formin). These mutations are more commonly found in infants, young (<25 years of age) patients who do not respond to glucocorticoid therapy, and patients with a family history of ESKD. Individuals of African ancestry carry approximately a five times higher risk of FSGS than those of European descent, likely mediated in large part by variants in the APOL1 gene. The third major cause of podocyte injury, hyperfiltration, causes a large proportion of secondary FSGS cases. This hyperfiltration form of FSGS is seen classically in patients with obesity but also can manifest in patients with a history of premature birth or solitary kidney. Finally, a drug-induced FSGS can occur as a rare complication of some commonly used treatments, including lithium, interferon, and pamidronate.
Associated with HIV, heroin, obesity, african american, sickle cells
Clinical Manifestations
All patients with FSGS have proteinuria. Primary forms of FSGS usually present with nephrotic-range proteinuria and can be accompanied by the full nephrotic syndrome, including severe edema, whereas secondary forms typically have asymptomatic subnephrotic proteinuria. Any form of FSGS can present with reduced glomerular filtration rate, particularly when diagnosed late in the disease course.
Diagnosis
FSGS is diagnosed by kidney biopsy. On light microscopy, glomerulosclerosis (scarring of glomeruli) is seen in a focal (<50%) and segmental (affecting a portion but not the entirety of individual glomeruli) distribution (Figure 12). Histologic variants of FSGS include classic (or “not otherwise specified”) FSGS, perihilar variant, glomerular tip lesion variant, cellular variant, and collapsing variant. The collapsing variant is often resistant to treatment and therefore carries the worst long-term prognosis, whereas the tip lesion usually responds to glucocorticoids and is unlikely to progress to ESKD.
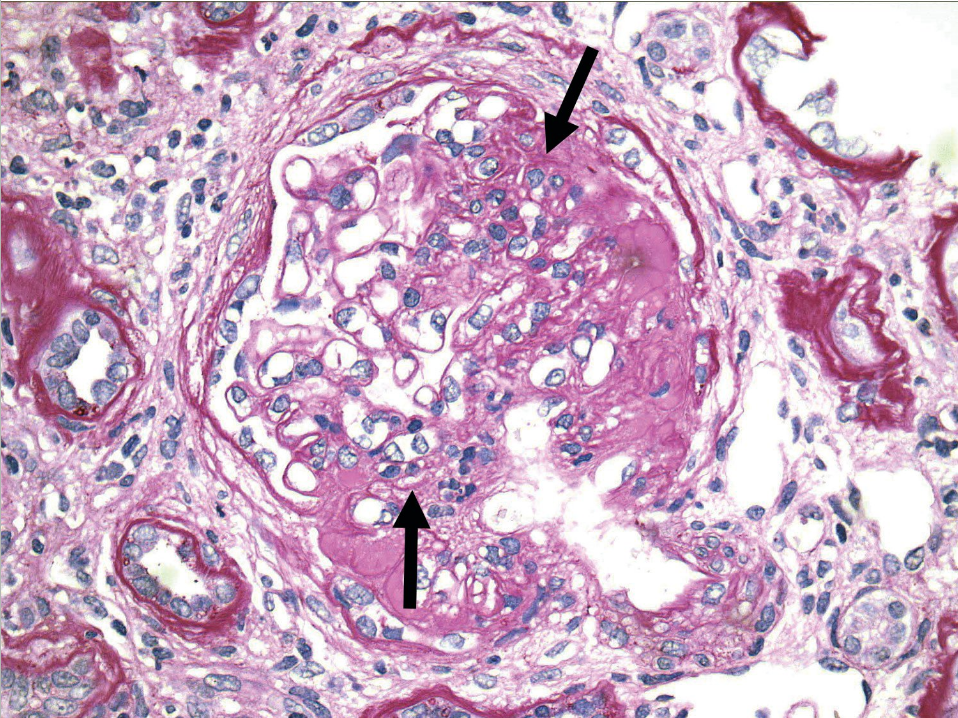 Light microscopy in focal segmental glomerulosclerosis shows scarring (arrows) in some, but not all, glomeruli in a segmental distribution, as demonstrated by this glomerulus.
Light microscopy in focal segmental glomerulosclerosis shows scarring (arrows) in some, but not all, glomeruli in a segmental distribution, as demonstrated by this glomerulus.
Treatment and Prognosis
For all patients with FSGS, conservative therapy includes renin-angiotensin system blocking drugs to control proteinuria, cholesterol-lowering medication, and edema management. Such conservative therapy measures suffice for FSGS patients with subnephrotic proteinuria.
When nephrotic-range proteinuria is present and if a primary form of FSGS is suspected, high-dose glucocorticoids remain the mainstay of initial therapy. Other immunosuppressants, including calcineurin inhibitors, mycophenolate mofetil, alkylating agents, and rituximab, have been reported to provide some benefit as second-line agents for patients who either do not respond to glucocorticoids or whose proteinuria relapses when glucocorticoids are tapered off.
Approximately 50% of patients with FSGS progress to ESKD within 10 years of diagnosis; this poor prognosis is rooted in low rates of treatment response.