CAH
Congenital adrenal hyperplasia (aka CAH or adrenogenital syndrome) is an autosomal recessive disorder caused by deficiencies in adrenal steroid synthesis due to enzymatic defects.
The three main enzymatic deficiencies are:
- 21-OH
- 11-OH
- 17-OH
The most common cause of CAH is 21-hydroxylase deficiency.
There is no negative feedback due to the deficient adrenal steroid synthesis. Therefore, there will be increased ACTH production, which leads to adrenal hyperplasia (including androgen excess)
A unifying feature between 21, 11, and 17-OH deficiencies is that a deficiency in one pathway causes precursor shunting to the remaining functional pathways
So, an enzyme deficiency in one pathway may lead to overproduction of a different enzyme in another pathway.
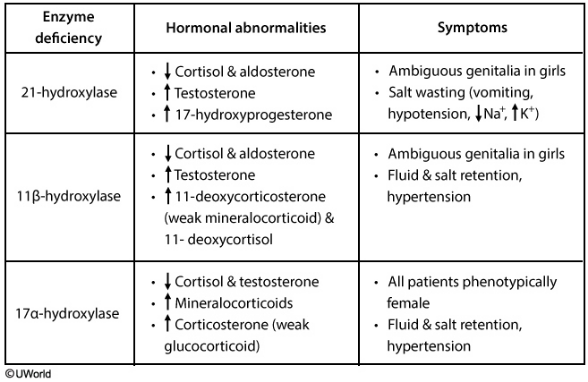
21-OH
21-OH deficiency: This enzyme is responsible for the synthesis of aldosterone and cortisol.
Deficiency leads to:
- ↓ Aldosterone
- ↓ Cortisol
- ↑ Androgens
Clinically, this presents as salt wasting with hypotension and ambiguous female genitalia, or male precocious puberty.
Labs:
- Hyponatremia
- Hyperkalemia
- Excess androgen
11-OH
11-OH deficiency:
This enzyme is one step down the cortisol/aldosterone synthesis pathway. As such, it presents similarly to 21-OH deficiency, with one key difference:
The aldosterone precursor that accumulated in a 21-OH deficiency didn’t have any hormonal activity, but a deficiency in 11-hydroxyase allows for production of 11-deoxycorticosterone, which has a weak mineralocorticoid effect. This causes salt and water retention.
Therefore, clinically, it presents with hypertension and ambiguous female genitalia or precocious male puberty (from excess androgens).
21 and 11 are similar numbers, so remember that they are in the same pathway and cause similar effects. Additionally, 11-OH deficiency can be remembered with the mnemonic “↑↑-OH deficiency” with up arrows representing the hypertension and masculinized genitalia.
17-OH
17-OH deficiency:
Deficiency in this enzyme prevents androgens and cortisol production. This results in high aldosterone levels and dysfunctional sexual development.
Males will be born with either feminized or completely female external genitalia and hypertension (from salt and water retention)
Since female genitalia is the default pathway, females will appear sexually normal at birth. However, since androgens are essential to puberty, the females will present with a lack of secondary sexual development and amenorrhea. Like males, they will also have hypertension.
Labs:
- Hypernatremia --> HTN
- Hypokalemia
- Decreased androgens
Diagnostic Workup:
- Thorough History and Physical exam including genitalia
- Blood pressure
- Basic Metabolic Panel (Na, K)
Imaging studies may also help with the diagnosis
-
Abdominal ultrasound to evaluate the adrenal glands
-
Ultrasound can also be used to diagnose testicular adrenal rest tumors (benign testicular tumors found in both classic and non-classic CAH) and should be done beginning in adolescence
Classic CAH
Classic CAH (impaired cortisol and aldosterone) may cause adrenal crisis in the newborn, if severe. In the salt-wasting form, newborn infants develop symptoms shortly after birth that include:
- Vomiting
- Dehydration
- Electrolyte imbalances
- Cardiac arrhythmia
Newborn screening for 21-hydroxylase deficiency is now a part of many screening programs. The initial evaluation would consist of blood chemistry to look at the kidney function and potassium level as well as renin and aldosterone levels.
Further evaluation of CAH involves the measurement of specific hormonal levels.
- For 21-OH deficiency, measuring 17-hydroxyprogesterone levels is important for diagnosis. The levels are elevated in this condition.
- In 11-OH deficiency, there are elevated levels of 11-deoxycortisol (or 11 DOC for short).
- An ACTH stimulation test is used to confirm the diagnosis and the type of CAH.
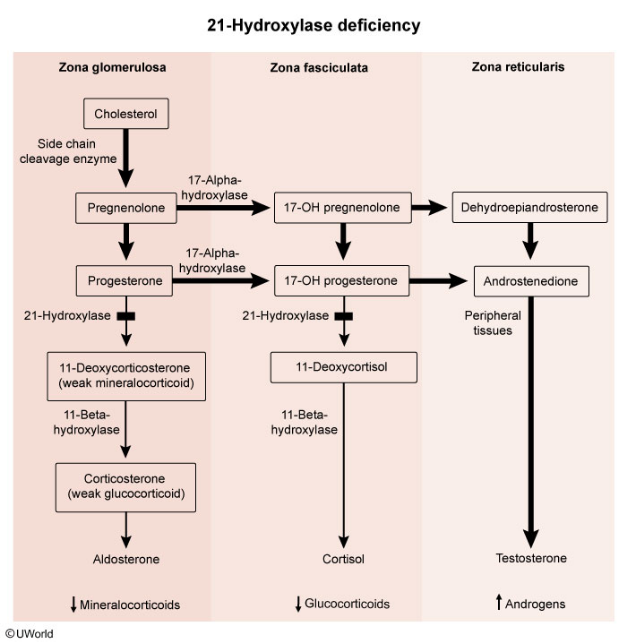
Marked elevation of 17-hydroxyprogesterone, a substrate of 21-hydroxylase, confirms the diagnosis with 21-hydroxylase deficiency. High testosterone levels are responsible for virilization of external female genitalia. Aldosterone and cortisol levels are abnormally low.
Treatment
The mainstay of treatment for patients with congenital adrenal hyperplasia is exogenous glucocorticoids and mineralocorticoids (e.g. fludrocortisone).
The first-line glucocorticoid depends on the age of the patient; in children, hydrocortisone is used due to its short half life, which decreases the risk of iatrogenic short stature. In adults, dexamethasone is used.
Stress doses of glucocorticoids should be used in patients with congenital adrenal hyperplasia that are experiencing physical stress or illness to avoid an adrenal crisis.